Prior to that, however, the FDA had already announced its intention to issue a proposed rule giving the agency regulatory oversight of LDTs.
“The FDA has continually supported the passage of the VALID Act by Congress,” attorney Charles Dunham IV, a Shareholder at Greenberg Traurig LLP in Houston, told Dark Daily. “In fact, there is speculation that the VALID Act will be attached to the Pandemic and All Hazards Preparedness Act as it moves through Congress.”
“The FDA may not actually proceed with promulgating rules to regulate LDTs if it is concerned it will not be successful in court if the rules are challenged, which would happen,” said attorney Charles Dunham IV (above), a Shareholder at Greenberg Traurig, LLP. Clinical laboratory leaders can learn more from Dunham during a panel discussion at next week’s 2023 Executive War College on Diagnostics, Clinical Laboratory, and Pathology Management in New Orleans. (Photo copyright: Greenberg Traurig LLP.)
Arguments For and Against FDA LDT Regulation of LDTs
Supporters of the VALID Act contend that putting LDTs under FDA regulation will lead to improved patient safety and less review for low-risk tests. Their argument is that LDTs should undergo the same FDA review and approval process as other medical devices.
Hospital laboratory managers and pathologists—particularly in academic medical center laboratories—have largely opposed FDA regulation of LDTs. They prefer to keep the current setup under which lab-developed tests are validated under the Clinical Laboratory Improvement Amendments of 1988 (CLIA). They argue that FDA intervention will slow down development of new tests.
In response, an FDA official indicated during the American Clinical Laboratory Association’s (ACLA) annual meeting on March 1 that the federal agency plans to issue a proposed rule to regulate LDTs, BioWorld reported. That rulemaking has not yet emerged. It’s possible the FDA will wait and see what happens in Congress with the VALID Act.
“Some legal experts have suggested that one significant new legal challenge FDA may face is the Supreme Court’s West Virginia v. EPA decision last summer that limited the ability of the EPA to cap power plant emissions by regulation due to the EPA’s lack of explicit congressional authority to do so,” said Gee, who also will appear on the Executive War College legal panel next week.
“The West Virginia v. EPA ruling provides support for those in the clinical lab industry who point to the FDA’s lack of clear statutory authority to regulate LDTs and therefore fundamentally disagree with FDA’s longstanding position that LDTs are medical devices subject to FDA’s authority to regulate,” he added.
Actions Clinical Laboratory Managers Can Take
Clinical laboratory managers who want to share their thoughts about the future of LDT regulation may want to take one or both of the following actions:
Contact their representatives in Congress.
Find out whether any trade associations they belong to have taken a position on the VALID Act.
Clinical laboratory professionals should monitor the VALID Act’s progress while also paying attention to industry groups and manufacturers that support or oppose the bill.
Doing so will provide a clearer indication of who has the most to gain or lose should the legislation be passed. Pathologists and medical laboratory managers should also remain alert for further efforts by the FDA to issue proposed rulemaking to regulate LDTs.
But even though the College of American Pathologists (CAP) and nine other organizations signed a December 12 stakeholder letter to leaders of key House and Senate committees urging passage of legislation that would enable some regulation of LDTs, the VALID Act was ultimately omitted from the year-end omnibus spending bill (H.R. 2617).
That may be due to pressure from organizations representing clinical laboratories and pathologists which lobbied hard against the bill.
Responding to criticism of its stance on FDA oversight of LDTs, in a May 2022 open letter posted on the organization’s website, anatomic pathologist and CAP president Emily Volk, MD, said “we at the CAP have an honest difference of opinion with some other respected laboratory organizations. … We believe the VALID Act is the only viable piece of legislation addressing the LDT issue. … the VALID Act contains many provisions that are similar to policy the CAP has advocated for regarding the regulation of laboratory tests since 2009. Importantly, the current version includes explicit protections for pathologists and our ability to practice medicine without infringement from the Food and Drug Administration (FDA).” (Photo copyright: College of American Pathologists.)
Organizations on Both Sides Brought Pressure to Bear on Legislators
The AAMC and AMP were especially influential, Bucshon told ProPublica. In addition to spending hefty sums on lobbying, AMP urged its members to contact legislators directly and provided talking points, ProPublica reported.
“The academic medical centers and big medical centers are in every state,” Bucshon said. As major employers in many locales, they have “a pretty big voice,” he added.
Discussing CAP’s reasoning behind its support of the VALID Act in a May 26 open letter and podcast, CAP president Emily Volk, MD, said the Valid Act “creates a risk-based system of oversight utilizing three tiers—low, moderate and high risk—in order to target the attention of the FDA oversight.”
While acknowledging that it had room for improvement, she lauded the bill’s three-tier risk-based system, in which tests deemed to have the greatest risks would receive the highest level of scrutiny.
She also noted that the bill exempts existing LDTs from an FDA premarket review “unless there is a safety concern for patients.” It would also exempt “low-volume tests, modified tests, manual interpretation tests, and humanitarian tests,” she wrote.
In addition, the bill would “direct the FDA not to create regulations that are duplicative of regulation under CLIA,” she noted, and “would require the FDA to conduct public hearings on LDT oversight.”
Pros and Cons of the VALID Act
One concern raised by opponents relates to how the VALID Act addressed user fees paid by clinical laboratories to fund FDA compliance activities. But Volk wrote that any specific fees “would need to be approved by Congress in a future FDA user fee authorization bill after years of public input.”
During the May 2022 podcast, Volk also cast CAP’s support as a matter of recognizing political realities.
“We understand that support for FDA oversight of laboratory-developed tests or IVCTs is present on both sides of the aisle and in both houses of Congress,” she said. “In fact, it enjoys wide support among very influential patient advocacy groups.” These groups “are very sophisticated in their understanding of the issues with laboratory-developed tests, and they do have the ear of Congress. There are many in the laboratory community that believe the VALID Act goes too far, but I can tell you that many of these patient groups don’t believe it goes far enough and are actively pushing for even more restrictive paradigms.”
Also urging passage of the bill were former FDA commissioners Scott Gottlieb, MD, and Mark B. McClellan, MD, PhD. In a Dec. 5 opinion piece for STAT, they noted that “diagnostic technologies have undergone considerable advances in recent decades, owing to innovation in fields like genomics, proteomics, and data science.” However, they wrote, laws governing FDA oversight “have not kept pace,” placing the agency in a position of regulating tests based on where they are made—in a medical laboratory or by a manufacturer—instead of their “distinctive complexity or potential risks.”
In their May 22 letter, opponents of the legislation outlined broad areas of concern. They contended that it would create “an onerous and complex system that would radically alter the way that laboratory testing is regulated to the detriment of patient care.” And even though existing tests would be largely exempted from oversight, “the utility of these tests would diminish over time as the VALID Act puts overly restrictive constraints on how they can be modified.”
CLIA Regulation of LDTs also Under Scrutiny
The provision to avoid duplication with the Clinical Laboratory Improvement Amendments (CLIA) program—which currently has some regulatory oversight of LDTs and IVCTs—is “insufficient,” opponents added, “especially when other aspects of the legislation call for requirements and activities that lead to duplicative and unnecessary regulatory burden.”
Opponents to the VALID Act also argued that the definitions of high-, medium-, and low-risk test categories lacked clarity, stating that “the newly created definition of moderate risk appears to overlap with the definition of high risk.”
The opponents also took issue with the degree of discretion that the bill grants to the US Secretary of Health and Human Services. This will create “an unpredictable regulatory process and ambiguities in the significance of the policy,” they wrote, while urging the Senate committee to “narrow the discretion so that stakeholders may better evaluate and understand the implications of this legislation.”
Decades ago, clinical laboratory researchers were allowed to develop assays in tandem with clinicians that were intended to provide accurate diagnoses, earlier detection of disease, and help guide selection of therapies. Since the 1990s, however, an industry of investor-funded laboratory companies have brought proprietary LDTs to the national market. Many recognize that this falls outside the government’s original intent for encouragement of laboratory-developed tests to begin with.
Proteins in human saliva make up its proteome and may be the key to new, precision medicine diagnostics that would give clinical pathologists new capabilities to identify disease
Clinical pathologists may soon have an array of new precision medicine diagnostic tools based on peoples’ saliva. There are an increasing number of “–omes” that can be the source of useful diagnostic biomarkers for developing clinical laboratory tests. The latest is the world’s first saliva protein biome wiki.
Called the Human Salivary Proteome Wiki (HSP Wiki), the “public data platform,” which was created by researchers at the University of Buffalo, is the “first of its kind,” according to Labroots, and “contains data on the many thousands of proteins present in saliva.”
The HSP Wiki brings together data from independent studies on proteins present in human saliva. One of the researchers’ goals is to speed up the development of saliva-based diagnostics and personalized medicine tools.
In “The Human Salivary Proteome Wiki: A Community-Driven Research Platform,” published in the Journal of Dental Research, the researchers wrote, “Saliva has become an attractive body fluid for on-site, remote, and real-time monitoring of oral and systemic health. At the same time, the scientific community needs a saliva-centered information platform that keeps pace with the rapid accumulation of new data and knowledge by annotating, refining, and updating the salivary proteome catalog.
“We developed the Human Salivary Proteome (HSP) Wiki as a public data platform for researching and retrieving custom-curated data and knowledge on the saliva proteome. … The HSP Wiki will pave the way for harnessing the full potential of the salivary proteome for diagnosis, risk prediction, therapy of oral and systemic diseases, and preparedness for emerging infectious diseases,” they concluded.
“This community-based data and knowledge base will pave the way to harness the full potential of the salivary proteome for diagnosis, risk prediction, and therapy for oral and systemic diseases, and increase preparedness for future emerging diseases and pandemics,” Stefan Ruhl, DDS, PhD (above right, with Omer Gokcumen, PhD, Associate Professor of Biological Sciences on left), Professor, Department of Oral Biology, University of Buffalo, and lead researcher of the study, told Labroots. Development of precision medicine clinical laboratory diagnostics is part of their research goals. (Photo copyright: University of Buffalo.)
Where Does Saliva Come From?
Saliva is a complex biological fluid that has long been linked to oral health and the health of the upper gastrointestinal tract. Only recently, though, have scientists begun to understand from where in the body saliva proteins originate.
The authors wrote: “Salivary proteins are essential for maintaining health in the oral cavity and proximal digestive tract, and they serve as potential diagnostic markers for monitoring human health and disease. However, their precise organ origins remain unclear.
“Through transcriptomic analysis of major adult and fetal salivary glands and integration with the saliva proteome, the blood plasma proteome, and transcriptomes of 28+ organs, we link human saliva proteins to their source, identify salivary-gland-specific genes, and uncover fetal- and adult-specific gene repertoires,” they added.
“Our results pave the way for future investigations into glandular biology and pathology, as well as saliva’s use as a diagnostic fluid,” the researchers concluded.
Saliva plays a crucial role in digestion by breaking down starches. It also provides a protective barrier in the mouth. When salivary glands malfunction, patients can face serious health consequences. Although clinicians and scientists have long understood the importance of saliva to good health, the question now is whether it contains markers of specific diseases.
“The Human Salivary Proteome Wiki contains proteomic, genomic, transcriptomic data, as well as data on the glycome, sugar molecules present on salivary glycoproteins. New data goes through an interdisciplinary team of curators, which ensures that all input data is accurate and scientifically sound,” noted Labroots.
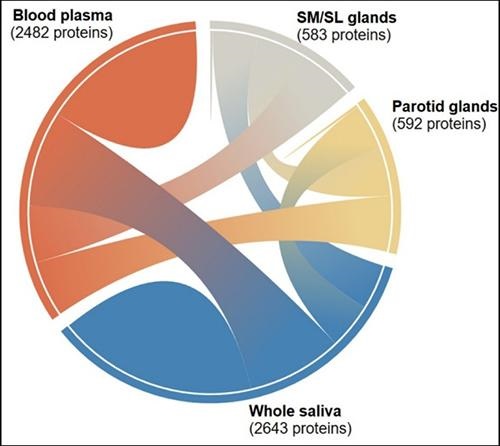
The graphic above “shows the interconnectedness of the thousands of salivary proteins originating from blood plasma, parotid glands, and submandibular and sublingual glands. The diagram is one of many tools available to researchers and clinicians through the Human Salivary Proteome Wiki,” noted a UBNow blog post. (Graphic copyright: University of Buffalo.)
Omics and Their Role in Clinical Laboratory Diagnostics
Proteomics is just one of several hotly-researched -omics that hold the potential to develop into important personalized medicine and diagnostics tools for pathologists. Genomics is a related area of research being studied for its potential to benefit precision medicine diagnostics.
However, unlike genomes, which do not change, proteomes change constantly. That is one of the main reasons studying the human salivary proteome could lead to valuable diagnostics tools.
Combining the study of the -omes with tools like mass spectrometry, a new era of pathology may be evolving. “With the rapid decrease in the costs of omics technologies over the past few years, whole-proteome profiling from tissue slides has become more accessible to diagnostic labs as a means of characterization of global protein expression patterns to evaluate the pathophysiology of diseases,” noted Pathology News.
Saliva and the Age of Precision Medicine
The study of the -omes may be an important element in the evolution of precision medicine, because of its ability to provide information about what is happening in patients’ bodies at the point of care.
Thus, a full understanding of the proteome of saliva and what causes it to change in response to different health conditions and diseases could open the door to an entirely new branch of diagnostics and laboratory medicine. It is easy and non-invasive to gather and, given that saliva contains so much information, it offers an avenue of study that may improve patients’ lives.
It also would bring us closer to the age of precision medicine where clinical laboratory scientists and pathologists can contribute even more value to referring physicians and their patients.
Though the potential is high for false positives and false negatives, some experts believe at-home COVID-19 testing still holds promise for slowing the spread of the coronavirus
The kit includes a nasal swab for specimen collection and a shipping package for returning the sample to a designated medical laboratory. Pixel is designed to work with LabCorp’s COVID-19 RT-PCR test, a real-time reverse transcription polymerase chain reaction (rRT-PCR) test that determines if an active SARS-CoV-2 coronavirus is present. The Pixel specimen-collection kit can be purchased for $119 on LabCorp’s website.
Presently, the Pixel kit is only available to healthcare workers and first responders who are symptomatic or who believe they may have been exposed to the virus. However, in a news release LabCorp stated that it “intends to make COVID-19 self-collection kits available to consumers in the coming weeks.”
Though purchasers have to pay for the kit themselves, a notice on LabCorp’s website states that the company “will work with you to get your purchase reimbursed by your health plan,” and that LabCorp is “actively working on a more streamlined solution, so you don’t have to pay up front.” LabCorp created a COVID-19 microsite where customers can receive future updates on the Pixel at-home test kit.
In LabCorp’s news release, Adam Schechter (at podium), President and CEO, emphasized his company’s commitment to helping patients and healthcare providers fight the COVID-19 crisis through LabCorp’s “leading testing capabilities and deep scientific and research expertise,” adding, “Our at-home collection kits are designed to make it easier and safer to test healthcare workers and first responders during this important time.” (Photo copyright: Yahoo News/Chip Somodevilla.)
Though Finger-stick At-home Tests Prove Inaccurate, Optimism Remains
As COVID-19 wreaks havoc around the globe, in vitro diagnostic (IVD) developers, clinical laboratory companies, and healthcare professionals have scrambled to find an accurate, cost effective way to definitively test individuals for the coronavirus.
Complicating matters is the fact that many people are asymptomatic carriers who show no symptoms of the illness, but who can infect others.
Earlier this year, the UK government was optimistic that an at-home serological antibodies test would enable its citizens to collect their own blood specimens via finger sticks, and that the test would provide a way for individuals to test themselves for the coronavirus.
According to CNBC, the United Kingdom (UK) ordered millions of antibody tests, but after disappointing results, returned the kits and requested a refund.
The New York Times (NYT) reported that the British government paid $20 million upfront for two million untried antibody test kits from two Chinese companies: AllTest Biotech in Hangzhou and Wondfo Biotech in Guangzhou. Then, UK government officials announced the tests would be available to citizens within weeks, and Prime Minister Boris Johnson publicly declared the tests would be “simple as a pregnancy test.”
Neither of those predictions would come to pass. In April, British researchers announced that none of the coronavirus tests they had tried were accurate enough to be of any value.
“We see many false negatives … and we also see false positives,” he wrote, adding that the UK “is now uniquely positioned to evaluate and find the optimal test for this disease, but no country has found a kit that is up to standard.” He also noted that locating such a test should be possible, but that it may take another month or more to find.
The Chinese companies defended their tests. In the Chinese newspaper Global Times, Wondfo stated its tests are “intended only as a supplement for patients who had already tested positive for the virus,” and on its website, AllTest stated its tests should “only [be] used by professionals,” not by patients at home, the New York Times reported.
Will At-home COVID-19 Testing Ever Work?
At-home testing kits for COVID-19 may seem like a great solution to the testing dilemma, but they could also prove to be problematic. “This may not be as good as it sounds,” Edo Paz, MD, a New York Presbyterian-trained cardiologist, Clinical Director at Heartbeat Health, and Vice President Medical, at K Health, a digital health company located in New York City, told CNET.
“Collecting a proper sample from the nose or mouth takes training and shipping delays of the specimen back to the lab could impact the quality of the sample,” he said, adding, “There could be a high false negative rate, leading people who are actually infected to believe they are not, potentially contributing to the crisis.”
Clinical pathologists have a unique understanding of the challenges that must be overcome for capillary blood to be of any use for testing, and of the potential for mishandling of specimens inherent in at-home test kits.
Nevertheless, with the SARS-CoV-2 coronavirus continuing to infect people around the world, the number and variety of tests will likely increase, which could create an upsurge in business for clinical laboratories and present new challenges for performing COVID-19 tests.
Lack of regulations and quality management jeopardizes the quality and safety of LDTs, claim experts in clinical laboratory medicine in a commentary to Canadian policymakers
The IHPME members published their comments in the Canadian Medical Association Journal (CMAJ), a peer-reviewed journal owned by Joule Inc., a subsidiary of the Canadian Medical Association. In it, they claim “recent expansion of the molecular diagnostics industry has revealed weaknesses in Canada’s regulatory system for laboratory-developed tests, which are not subject to statutory regulations on medical devices.”
For pathologists and clinical laboratory professionals in both Canada and the United States, these recent actions show the concerns many experts have as they watch the explosive growth in the use of laboratory-developed tests in both countries. In many ways, the swift advances in molecular and genetic diagnostics is outrunning the ability of government regulators to keep pace with use of LDTs in clinical care settings.
In their commentary in CMAJ, the IHPME members also
claim the review and evaluation of LDTs in Canada is inconsistent. Some LDTs they
say, may endure stringent assessments and have endorsements by clinical
guidelines or findings that are published in scientific journals. Other LDTs,
however, may have no analysis at all.
In addition, the IHPME members point out that there is no
national registry kept of LDTs. They theorize that a lack of proper regulation,
controls, and quality management “has potentially jeopardized the delivery of
quality, safe, timely, and appropriate care.”
The researchers calling on Health Canada to address these
issues include:
Fiona A. Miller, PhD, Professor of Health Policy and IHPME Chair in Health Management Strategies;
François Rousseau, PhD, Professor, Department of Molecular Biology, Medical Biochemistry and Pathology, Faculty of Medicine, Laval University, Quebec;
Alberto Gutierrez, PhD, Partner, NDA Partners LLC, former Director, Office of In Vitro Diagnostics and Radiological Health at the FDA’s Center for Devices and Radiological Health (CDRH);
Stuart Hogarth, PhD, Lecturer in Sociology of Science and Technology, University of Cambridge, Cambridge, UK.
During an exclusive presentation offered by The Dark Report (Dark Daily’s sister publication) in 2015, Alberto Gutierrez, PhD (above), who at that time was Director, Office of In Vitro Diagnostics and Radiological Health at the FDA, said, “LDTs are an area that will be difficult to regulate. There is a broad set of tests. Some of the LDTs are very good. Some of them require a lot of expertise from the pathologists and some of them don’t. Regulating LDTs in a way that makes sense and that does not disrupt what’s going on [in clinical laboratories] is going to be difficult.” (Photo copyright: FDA.)
Canadian Scientists Call on Health Canada to Take the
Lead on Regulating LDTs
In the US, the FDA has been making moves to regulate LDTs since 2010, with much opposition from clinical laboratories and In Vitro Diagnostic (IVD) manufacturers. The FDA describes LDTs as internally designed clinical laboratory tests that are developed, manufactured, and used within a single laboratory. They have not undergone government regulatory review, can be simple or complex, and can be utilized to detect a variety of analytes.
Health Canada is the name of a department that falls under
the purview of the Minister of
Health and is part of Canada’s Health
Portfolio. It is responsible for helping Canadians maintain and improve
their health. Other agencies included in the Health Portfolio are:
According to the IHPME paper, however, Health Canada
currently does not have a way to regulate LDTs, and no government agency in
that country is responsible for the oversight of laboratory-developed tests.
Only LDTs that are marketed as test kits are evaluated and reviewed by Health
Canada.
“The current laboratory regulatory system in Canada involves a mixture of public and private entities and operates with oversight from provincial governments, nongovernmental organizations, and professional societies,” the IHPME paper states, adding, “most provinces and territories rely on voluntary standards that are unevenly applied, with little auditing and systematic testing to ensure quality.”
The authors also note that the current lab regulations in
Canada apply only to the operations of the medical laboratories themselves,
encompassing such things as lab environments, personnel, accreditation, and
quality control. They believe the loophole regarding LDTs needs to be addressed,
and they urged Health Canada to “demonstrate leadership” by subjecting these
tests to regulations that are currently applied to medical devices and
pharmaceuticals.
Other Countries Regulate LDTs, though Not Without
Controversy
In support of their call to action, IHPME researchers noted
that Australia, the EU, and the US all have taken steps to regulate LDTs.
The Australian government began oversight of LDTs in 2010 by
subjecting high-risk LDTs to external evaluation and then tracking them in a
public registry.
An EU regulation, which was passed in 2017, will administer
regulatory review of LDTs manufactured on an industrial scale, which targets
commercial laboratories. The law exempts LDTs utilized within individual
hospital laboratories and should be fully implemented by 2022.
Though on its radar since the 1990s, in 2010, the FDA officially
announced its intent to regulate LDTs in the US. The agency released an initial
draft approach for doing so starting in 2014, held a public workshop on the
topic in 2015, and released a
discussion paper in 2017. At this time, however, the FDA is not regulating
LDTs, though the agency remains open to the possibility.
Dark Daily
has reported extensively over the years on the development of LDTs and the
controversy surrounding the FDA’s moves to regulate them.
According to the FDA
website, problems with several high-risk LDTs have been identified,
including:
Claims that are not adequately supported with
evidence;
Lack of appropriate controls which may yield
erroneous results; and
The FDA’s report, titled, “The
Public Health Evidence for FDA Oversight of Laboratory Developed Tests,” reviewed
20 case studies of LDTs for Lyme disease, ovarian cancer, whooping cough,
fibromyalgia, prostate cancer, autism, breast cancer, melanoma, Vitamin D, and
other conditions. The agency concluded that in many instances “patients have
been demonstrably harmed or may have been harmed by tests that did not meet FDA
requirements.”
Klein noted, however, that “The 20 tests described by FDA are mostly a hodgepodge of outlier assays including tests that were never offered, tests for which comparable FDA assays perform poorly, tests for poorly defined disorders with psychologic components, and use of an FDA-approved test off-label.” He continued, “That FDA could find only these dubious examples out of the many thousands of laboratory-developed procedures (LDPs) that benefit patients each day, calls into question the agency’s rationale for expanding its regulatory scope to include LDPs.”
Perhaps this is why the FDA has yet to implement regulations
for LDTs. The controversy continues.
Whether Health Canada will accept the advice of the IHPME
scientists and take steps to regulate laboratory-developed tests in Canada remains
to be seen. As more LDTs are created and manufactured, however, it is probable
that governments will continue to evaluate the administration and oversight of laboratory-developed
tests.
In both Canada and the United States, pathologists, clinical
laboratory managers, and executives at in vitro diagnostic manufacturers
can expect an ongoing tug-of-war between government regulators and the lab
industry over the most appropriate ways to regulate LDTs.