


Prepare Your Playbook to Obtain FDA Approval for Your Laboratory Developed Tests
Presented by: Sheila Walcoff, Valerie Palmieri, Jane Pine Wood Prepare Your Playbook to Obtain FDA Approval for Your Laboratory Developed Tests by Sheila Walcoff, Valerie Palmieri, Jane Pine Wood https://www.darkdaily.com/wp-content/uploads/Audio%20Files/2024EWC/V-Palmieri-J-Wood-S-Walcoff-245-Foster1.mp3 PDF copy of the Presentation Slides...Former FDA Director to Speak at Executive War College on FDA’s Coming Regulation of Laboratory Developed Tests
Tim Stenzel, MD, PhD, will discuss what clinical laboratories need to know about the draft LDT rule, FDA memo on assay reclassification, and ISO-13485 harmonization
Many clinical laboratories anxiously await a final rule from the US Food and Drug Administration (FDA) that is expected to establish federal policies under which the agency will regulate laboratory developed tests (LDTs). The agency released a proposed rule on Oct. 3, 2023, setting a Dec. 4 deadline for submission of comments. The White House’s Office of Management and Budget received a draft of the final rule less than three months later on March 1, 2024.
“Given how fast it moved through HHS, the final [rule] is likely pretty close” to the draft version, wrote former FDA commissioner Scott Gottlieb, MD, in a post on LinkedIn. Gottlieb and other regulatory experts expect the White House to submit the final rule to Congress no later than May 22, and perhaps as soon as this month.
But what will the final rule look like? Tim Stenzel, MD, PhD, former director of the FDA’s Office of In Vitro Diagnostics, suggests that it is too soon to tell.
Stenzel, who retired from the FDA last year, emphasized that he was not speaking on behalf of the federal agency and that he adheres to all FDA confidentiality requirements. He formed a new company—Grey Haven LLC—through which he is accepting speaking engagements in what he describes as a public service.

“I’m taking a wait and see approach,” said Tim Stenzel, MD, PhD (above), former director of the FDA’s Office of In Vitro Diagnostics, in an interview with Dark Daily. “The rule is not finalized. The FDA received thousands of comments. It’s my impression that the FDA takes those comments seriously. Until the rule is published, we don’t know what it will say, so I don’t think it does any good to make assumptions.” Clinical laboratory leaders will have an opportunity to learn how to prepare for FDA regulation of LDTs directly from Stenzel at the upcoming Executive War College in May. (Photo copyright: LinkedIn.)
FDA’s History of LDT Regulation
Prior to his five-year stint at the agency, Stenzel held high-level positions at diagnostics manufacturers Invivoscribe, Quidel Corporation, Asuragen, and Abbott Laboratories. He also directed the clinical molecular diagnostics laboratory at Duke University Medical Center in North Carolina. In the latter role, during the late 1990s, he oversaw development of numerous LDTs, he said.
The FDA, he observed, has long taken the position that it has authority to regulate LDTs. However, since the 1970s, after Congress passed the Medical Device Amendments to the federal Food, Drug, and Cosmetic Act, the agency has generally exercised “enforcement discretion,” he said, in which it declined to regulate most of these tests.
At the time, “many LDTs were lower risk, small volume, and used for specialized needs of a local patient population,” the agency stated in a press release announcing the proposed rule. “Since then, due to changes in business practices and increasing ability to ship patient specimens across the country quickly, many LDTs are now used more widely, for a larger and more diverse population, with large laboratories accepting specimens from across the country.”
Clinical Labs Need a Plan for Submission of LDTs to FDA
The FDA proposed the new rule after Congress failed to vote on the VALID Act (Verifying Accurate Leading-edge IVCT Development Act of 2021), which would have established a statutory framework for FDA oversight of LDTs. Citing public comments from FDA officials, Stenzel believes the agency would have preferred the legislative approach. But when that failed, “they thought they needed to act, which left them with the rulemaking path,” he said.
The new rule, as proposed, would phase out enforcement discretion in five stages over four years, he noted. Labs would have to begin submitting high-risk tests for premarket review about three-and-a-half years from publication of the final rule, but not before Oct. 1, 2027. Premarket review requirements for moderate- or low-risk tests would follow about six months later.
While he suggested a “wait and see” approach to the final rule, he advises labs that might be affected to develop a plan for dealing with it.
Potential Lawsuits
Stenzel also noted the likelihood of litigation in which labs or other stakeholders will seek to block implementation of the rule. “It’s a fairly widespread belief that there will be a lawsuit or lawsuits that will take this issue through the courts,” he said. “That could take several years. There is no guarantee that the courts will ultimately side with the FDA.”
In “Perfect Storm of Clinical Lab and Pathology Practice Regulatory Changes to Be Featured in Discussions at 29th Annual Executive War College,” Dark Daily covers how the forces in play will directly impact the operations and financial stability of many of the nation’s clinical laboratories.
Stenzel is scheduled to speak about the LDT rule during three sessions at the upcoming Executive War College on Diagnostic, Clinical Laboratory, and Pathology Management conference taking place on April 30-May 1 in New Orleans.
He acknowledged that it is a controversial issue among clinical laboratories. Many labs have voiced opposition to the rule as well as the Valid Act.
Currently in retirement, Stenzel says he is making himself available as a resource through public speaking for laboratory professionals and other test developers who are seeking insights about the agency.
“The potential value that I bring is recent experience with the FDA and with stakeholders both inside and outside the FDA,” he said, adding that during his presentations he likes “to leave plenty of time for open-ended questions.”
In the case of his talks at the Executive War College, Stenzel said he anticipates “a robust conversation.”
He also expects to address other FDA-related issues, including:
- A recent memo in which the agency said it would begin reclassifying most high-risk In Vitro Diagnostic (IVD) tests—those in class III (high risk)—into class II (moderate to high risk).
- The emergence of multi-cancer detection (MCD) tests, which he described as a “hot topic in the LDT world.” The FDA has not yet approved any MCD tests, but some are available as LDTs.
- A new voluntary pilot program in which the FDA will evaluate LDTs in situations where the agency has approved a treatment but has not authorized a corresponding companion diagnostic.
- An FDA effort to harmonize ISO 13485—a set of international standards governing development of medical devices and diagnostics—with the agency’s own quality system regulations. Compliance with the ISO standards is necessary to market products in many countries outside the US, particularly in Europe, Stenzel noted. Harmonization will simplify product development, he said, because manufacturers won’t have to follow two or more sets of rules.
To learn how to prepare for the FDA’s future regulation of LDTs, clinical laboratory and pathology group managers would be wise to attend Stenzel’s presentations at this year’s Executive War College. Visit here to learn more and to secure your seat in New Orleans.
—Stephen Beale
Related Information:
FDA Proposes Rule Aimed at Helping to Ensure Safety and Effectiveness of Laboratory Developed Tests
Proposed Rule Webinar: Medical Devices; Laboratory Developed Tests (webinar transcript)
Proposed Rule Webinar: Medical Devices; Laboratory Developed Tests (slides)
FDA Proposed Rule on Medical Devices; Laboratory Developed Tests
CDRH Announces Intent to Initiate the Reclassification Process for Most High Risk IVDs
Questions and Answers about Multi-Cancer Detection Tests Oncology Drug Products Used with Certain In Vitro Diagnostics Pilot Program
Survey: Proposed FDA Approval of Laboratory Developed Tests Will Reduce Innovation
Though response was limited in Dark Daily’s poll, the message from respondents was overwhelmingly negative on LDT regulation by the FDA
Most respondents to a recent industry survey said that should Food and Drug Administration (FDA) approval be required in the future for laboratory developed tests (LDTs), innovation will suffer.
This conclusion echoes what opponents of the Verifying Accurate Leading-Edge IVCT Development Act (VALID Act) have argued will happen should that proposed bill become law.
In the survey, which was conducted by Dark Daily, 91% of respondents said FDA pre-market approval of LDTs would decrease clinical laboratories’ ability to bring innovative tests to market. (See Figure 1.) The other 9% felt it would have no effect on innovation. Zero of the respondents said FDA involvement would increase innovative tests.
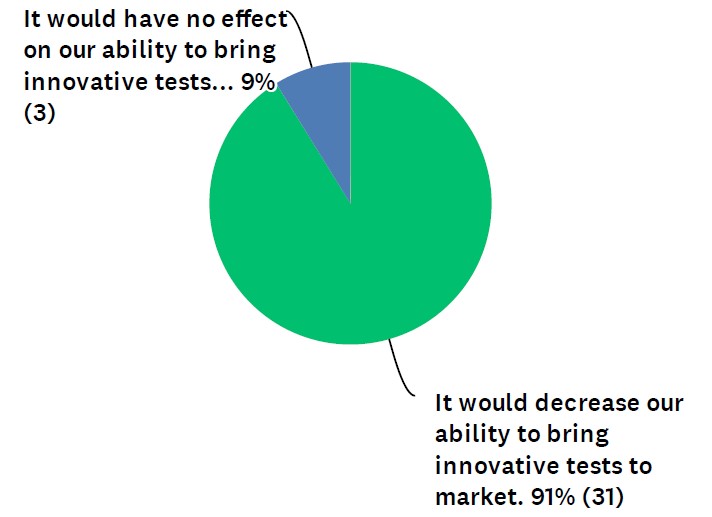
“Development of LDT tests has been the mission of most of the labs, and it meets the need for patient care,” noted one respondent in the survey. “Moving LDTs under FDA will create more obstacles for labs to offer the tests.”
To be fair, the survey had limited responses—34 in total. The poll went out to thousands of Dark Daily readers. We found that response rate surprising given how many labs will be affected if the VALID Act becomes law.
An LDT is a proprietary diagnostic test developed and performed by an individual clinical laboratory. In academic medical center laboratories, LDTs often address unmet clinical needs. The Clinical Laboratory Improvement Amendments of 1988 (CLIA) generally regulates LDTs.
VALID Act vs. VITAL Act Hinges on LDT Oversight
The VALID Act is a bipartisan bill that proposes FDA oversight of laboratory developed tests. The bill continues to make its way through the U.S. Senate and the House of Representatives.
A counterproposal called the Verified Innovative Testing in American Laboratories Act (VITAL Act) is also before Congress but has less momentum behind it. The VITAL Act seeks to keep LDTs under CLIA while also calling for reforms to account for modern lab tests.
See our past reporting in Dark Daily for a detailed comparison of the VALID Act and VITAL Act.
Looking at survey results, 80% “strongly disagreed” or “disagreed” that new LDT requirements, such as those found in the VALID Act, are needed. (See Figure 2.)
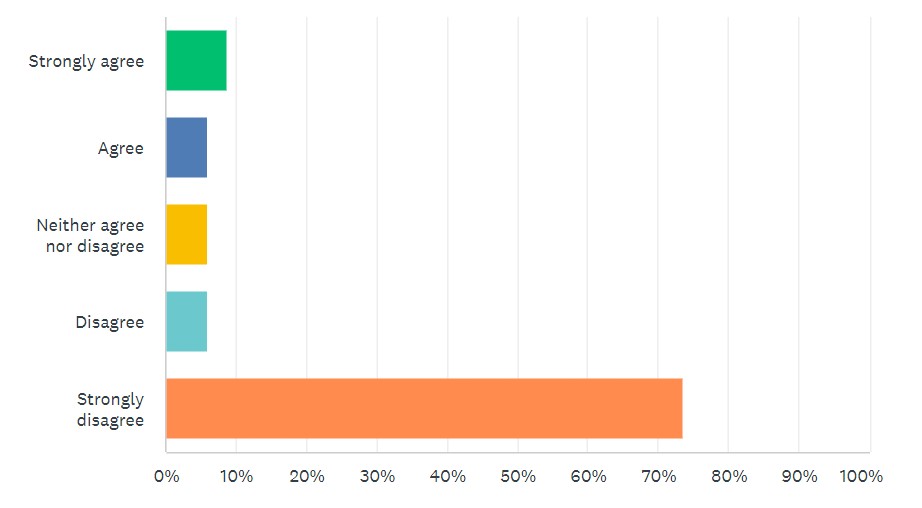
By comparison, 65% “strongly agreed” or “agreed” with modernizing CLIA requirements for LDTs, as called for in the VITAL Act.
Those numbers shifted somewhat depending on the lab setting of the respondent. For example, just looking at commercial labs, opposition to the VALID Act remained similar, but support for modernizing CLIA jumped up to 88%. When looking at just hospitals, independent labs, and academic labs, numbers for both topics remained consistent.
When filtering the answers, the number of lab employees in a setting had little effect on survey results.
Political Battle Continues Over Laboratory Developed Tests
Clinical laboratory industry groups and others have been amassing to oppose or support the VALID Act. For example, the Advanced Medical Technology Association and The Pew Charitable Trusts are behind the bill.
However, the American Association for Clinical Chemistry, Association for Molecular Pathology, and new Coalition for Innovative Laboratory Testing are against the VALID Act.
Congress may attach the VALID Act to the authorization vote for the Medical Device User Fee Agreement V (MDUFA). At this point, discussions on MDUFA remain in congressional committees.
—Scott Wallask
Related Information:
What is a laboratory developed test?
LDT Regulation: New Survey Asks Readers for Their Views About Two Bills Before Congress
MDUFA V Agreement Enables Bold MedTech Vision to Become Reality
Clinical Trial Shows New Laboratory Developed Blood Test 83% Effective at Detecting Colorectal Cancer
Accurate blood-based clinical laboratory testing for cancer promises to encourage more people to undergo early screening for deadly diseases
One holy grail in diagnostics is to develop less-invasive specimen types when screening or testing for different cancers. This is the motivation behind the creation of a new assay for colorectal (colon) cancer that uses a blood sample and that could be offered by clinical laboratories. The data on this assay and its performance was featured in a recent issue of the New England Journal of Medicine(NEJM).
The company developing this new test recognized that more than 50,000 people will die in 2024 from colon cancer, according to the American Cancer Society. That’s primarily because people do not like colonoscopies even though the procedure can detect cancer in its early stages. Similarly, patients tend to find collecting their own fecal samples for colon cancer screening tests to be unpleasant.
But the clinical laboratory blood test for cancer screening developed by Guardant Health may make diagnosing the deadly disease less invasive and save lives. The test “detects 83% of people with colorectal cancer with specificity of 90%,” a company press release noted.
“Early detection could prevent more than 90% of colorectal cancer-related deaths, yet more than one third of the screening-eligible population is not up to date with screening despite multiple available tests. A blood-based test has the potential to improve screening adherence, detect colorectal cancer earlier, and reduce colorectal cancer-related mortality,” the study authors wrote in the NEJM.
As noted above, this is the latest example of test developers working to develop clinical laboratory tests that are less invasive for patients, while equaling or exceeding the sensitivity and specificity of existing diagnostic assays for certain health conditions.

“I do think having a blood draw versus undergoing an invasive test will reach more people, My hope is that with more tools we can reach more people,” Barbara H. Jung, MD (above), President of the American Gastroenterological Association, told NPR. Clinical laboratory blood tests for cancer may encourage people who do not like colonoscopies to get regular screening. (Photo copyright: American Gastroenterology Association.)
Developing the Shield Blood Test
Colorectal cancer is the “third most common cancer among men and women in the US,” according to the American Gastrological Association (AGA). And yet, millions of people do not get regular screening for the disease.
To prove their Shield blood test, Guardant Health, a precision oncology company based in Redwood City, Calif., enrolled more than 20,000 patients between the ages of 45-84 from across the US in a prospective, multi-site registrational study called ECLIPSE (Evaluation of ctDNA LUNAR Assay In an Average Patient Screening Episode).
“We assessed the performance characteristics of a cell-free DNA (cfDNA) blood-based test in a population eligible for colorectal cancer screening. The coprimary outcomes were sensitivity for colorectal cancer and specificity for advanced neoplasia (colorectal cancer or advanced precancerous lesions) relative to screening colonoscopy. The secondary outcome was sensitivity to detect advanced precancerous lesions,” the study authors wrote in the NEJM.
In March, Guardant completed clinical trials of its Shield blood test for detecting colorectal cancer (CRC) in men and women. According to the company press release, the test demonstrated:
- 83% sensitivity in detecting individuals with CRC.
- 88% sensitivity in detecting pathology-confirmed Stages I-III.
Additionally, the Shield test showed sensitivity by stage of:
- 65% for pathology-confirmed Stage I,
- 55% for clinical Stage I,
- 100% for Stage II, and
- 100% for Stage III.
“The results of the study are a promising step toward developing more convenient tools to detect colorectal cancer early while it is more easily treated,” said molecular biologist and gastroenterologist William M. Grady, MD, Medical Director, Gastrointestinal Cancer Prevention Program at Fred Hutchinson Cancer Center and corresponding author of the ECLIPSE study in the press release. “The test, which has an accuracy rate for colon cancer detection similar to stool tests used for early detection of cancer, could offer an alternative for patients who may otherwise decline current screening options.”
Are Colonoscopies Still Needed?
“More than three out of four Americans who die from colorectal cancer are not up to date with their recommended screening, highlighting the need for a more convenient and less invasive screening method that can overcome barriers associated with traditional options,” Daniel Chung, MD, gastroenterologist at Massachusetts General Hospital and Professor of Medicine at Harvard Medical School, said in the Guardant press release.
Barbara H. Jung, MD, President of the American Gastroenterological Association, says that even if Guardant’s Shield test makes it to the public the “dreaded colonoscopy” will still be needed because the procedure is used to locate and test polyps. “And when you find those you can also remove them, which in turn prevents the cancer from forming,” she told NPR.
There is hope that less invasive clinical laboratory testing will encourage more individuals to get screened for cancer earlier and regularly, and that the shift will result in a reduction in cancer rates.
“Colorectal cancer is highly treatable if caught in the early stages,” said Chris Evans, President of the Colon Cancer Coalition, in the Guardant press release.
Guardant Health’s ECLIPSE study is a prime example of the push clinical laboratory test developers are making to create user-friendly test options that make it easier for patients to follow through with regular screening for early detection of diseases. It echoes a larger effort in the medical community to think outside the box and come up with creative solutions to reach wider audiences in the name of prevention.
—Kristin Althea O’Connor
Related Information:
A Cell-free DNA Blood-Based Test for Colorectal Cancer Screening
A Simple Blood Test Can Detect Colorectal Cancer Early, Study Finds
Key Statistics for Colorectal Cancer
