Prior to that, however, the FDA had already announced its intention to issue a proposed rule giving the agency regulatory oversight of LDTs.
“The FDA has continually supported the passage of the VALID Act by Congress,” attorney Charles Dunham IV, a Shareholder at Greenberg Traurig LLP in Houston, told Dark Daily. “In fact, there is speculation that the VALID Act will be attached to the Pandemic and All Hazards Preparedness Act as it moves through Congress.”
“The FDA may not actually proceed with promulgating rules to regulate LDTs if it is concerned it will not be successful in court if the rules are challenged, which would happen,” said attorney Charles Dunham IV (above), a Shareholder at Greenberg Traurig, LLP. Clinical laboratory leaders can learn more from Dunham during a panel discussion at next week’s 2023 Executive War College on Diagnostics, Clinical Laboratory, and Pathology Management in New Orleans. (Photo copyright: Greenberg Traurig LLP.)
Arguments For and Against FDA LDT Regulation of LDTs
Supporters of the VALID Act contend that putting LDTs under FDA regulation will lead to improved patient safety and less review for low-risk tests. Their argument is that LDTs should undergo the same FDA review and approval process as other medical devices.
Hospital laboratory managers and pathologists—particularly in academic medical center laboratories—have largely opposed FDA regulation of LDTs. They prefer to keep the current setup under which lab-developed tests are validated under the Clinical Laboratory Improvement Amendments of 1988 (CLIA). They argue that FDA intervention will slow down development of new tests.
In response, an FDA official indicated during the American Clinical Laboratory Association’s (ACLA) annual meeting on March 1 that the federal agency plans to issue a proposed rule to regulate LDTs, BioWorld reported. That rulemaking has not yet emerged. It’s possible the FDA will wait and see what happens in Congress with the VALID Act.
“Some legal experts have suggested that one significant new legal challenge FDA may face is the Supreme Court’s West Virginia v. EPA decision last summer that limited the ability of the EPA to cap power plant emissions by regulation due to the EPA’s lack of explicit congressional authority to do so,” said Gee, who also will appear on the Executive War College legal panel next week.
“The West Virginia v. EPA ruling provides support for those in the clinical lab industry who point to the FDA’s lack of clear statutory authority to regulate LDTs and therefore fundamentally disagree with FDA’s longstanding position that LDTs are medical devices subject to FDA’s authority to regulate,” he added.
Actions Clinical Laboratory Managers Can Take
Clinical laboratory managers who want to share their thoughts about the future of LDT regulation may want to take one or both of the following actions:
Contact their representatives in Congress.
Find out whether any trade associations they belong to have taken a position on the VALID Act.
Clinical laboratory professionals should monitor the VALID Act’s progress while also paying attention to industry groups and manufacturers that support or oppose the bill.
Doing so will provide a clearer indication of who has the most to gain or lose should the legislation be passed. Pathologists and medical laboratory managers should also remain alert for further efforts by the FDA to issue proposed rulemaking to regulate LDTs.
Organizations representing clinical laboratories and other critical healthcare providers urged Congress to pass the Saving Access to Laboratory Services Act by January 1, 2023, to prevent deep cuts in reimbursements
Lessons about the essential role of clinical laboratories during a pandemic was the central theme in a significant publication released recently. The authors were the presidents of two of the nation’s largest healthcare companies and their goal was to connect the value clinical labs delivered during the COVID-19 pandemic to the financial threat labs face should the Protecting Access to Medicare Act of 2014 (PAMA) fee cuts coming to the Medicare Part B Clinical Laboratory Fee Schedule (CLFS) be implemented.
In an article for RealClearPolicy, healthcare executives William G. Morice II, MD, PhD (left), CEO/President, Mayo Clinic Laboratories, and Matt Sause (right), President of Roche Diagnostics North America wrote, “Without PAMA reform, labs could face drastically reduced reimbursement for commonly performed lab tests for a host of diseases.” (Photo copyrights: Mayo Clinic Laboratories/Roche Diagnostics.)
IVD Companies and Clinical Laboratories Sound Alarm
Morice and Sause warn that—without PAMA reform—the nation’s vital medical laboratories will face “drastically reduced reimbursement” for commonly performed lab tests for diseases, including diabetes, heart disease, and cancer. Reimbursement cuts may cause clinical labs serving “the most vulnerable and homebound” to reduce services or close, they noted.
“To emerge from nearly three years of a pandemic by sending the signal that austerity is our nation’s health policy when it comes to testing and diagnostics would be a significant mistake,” they wrote.
“If the proposed cuts to reimbursements for diagnostic tests are allowed to take effect, disparities caused by challenges with accessing diagnostic tests will likely grow even further,” the authors continued.
However, they added, “The Saving Access to Laboratory Services Act [SALSA] would reform PAMA to require accurate and representative data from all laboratory segments that serve Medicare beneficiaries to be collected to support a commonsense Medicare fee schedule that truly represents the market.”
How PAMA Affects Clinical Laboratory Reimbursements
PAMA, which became law in 2014, was aimed at marrying Medicare Part B Clinical Laboratory Fee Schedule (CLFS) reimbursement rates to rates medical laboratories receive from private payers, the National Independent Laboratory Association (NILA) explained in a news release.
But from the start, in its implementation of the PAMA statute, the methods used by the federal Centers for Medicare and Medicaid Services (CMS) to collect data on lab test prices paid by private payers—which were the basis for calculating new lab test prices for the Medicare program—were criticized by many laboratory professionals and other health experts.
Critics frequently pointed out that several types of clinical laboratories were excluded from reporting their private payer lab test prices. Thus, the data collected and used by CMS did not accurately represent the true range of prices paid for clinical lab tests by private health insurance plans, said lab industry groups.
CMS regulations “exclude most hospital outreach laboratories and physician office laboratories from data collection. This approach depresses median prices and has led to deep cuts to lab reimbursement. Many tests were cut up to 30% in 2018 when the new system went into effect,” the America Association for Clinical Chemistry (AACC) noted in a statement.
On September 8, just weeks after publication of the article authored by Morice and Sause, 26 organizations representing clinical laboratories and diagnostics manufacturers sent a letter to Congressional leaders. In it they described the financial impact on labs due to the current law’s omission of some outreach and physician office lab testing, and they urged the passage of the SALSA legislation.
“The significant under-sampling led to nearly $4 billion in cuts to those labs providing the most commonly ordered test services for Medicare beneficiaries,” the organizations wrote in their letter. “For context, the total CLFS spend for 2020 was only $8 billion.”
Reimbursement Cuts to Lab Tests are Coming if SASLA Not Passed
“Without Congressional action, beginning on Jan. 1, 2023, laboratories will face additional cuts of as much as 15% to some of the most commonly ordered laboratory tests,” the NILA said.
“Enactment of the Saving Access to Laboratory Services Act (SALSA/H.R. 8188/S.4449) is urgently needed this year, to allow laboratories to focus on providing timely, high quality clinical laboratory services for patients, continuing to innovate, and building the infrastructure necessary to protect the public health,” NILA added.
In an editorial she wrote for Clinical Lab Products, titled, “Be a Labvocate: Help Pass SALSA Legislation,” Kristina Martin, Clinical Pathology Operations Director, Department of Pathology, University of Michigan Medicine said, “The SALSA legislation provides a permanent, pragmatic approach to evaluating the CLFS, eliminating huge swings, either positive or negative as it pertains to Medicare reimbursement. It also allows for a more comprehensive evaluation of data to be collected from a broader sampling of laboratory sectors.”
Uses statistical sampling for widely available tests performed by a “representative pool of all clinical laboratory market segments.”
Introduces annual “guardrails” aimed at creating limits for reductions as well as increases in CLFS rates.
Excludes Medicaid managed care rates since they are not true “market rates.”
Gives labs the option to exclude mailed remittances from reporting if less than 10% of claims.
Eases clinical labs’ reporting requirements by changing data collection from three years to four.
Make Your Views Known
Proponents urge Congress to act on SALSA before the end of the year. Clinical laboratory leaders and pathologists who want to express their views on SALSA, test reimbursement, and the importance of access to medical laboratory testing can do so through Stop Lab Cuts.org. The website is sponsored by the ACLA.
Survey respondents can give their opinions about the proposed VALID and VITAL acts
Two bills are pending in Congress, and each is written to change the current regulatory scheme for laboratory-developed tests (LDTs) and in vitro clinical tests (IVCTs). The bills go by the acronyms of the VALID Act and VITAL Act. Many clinical laboratories offering LDTs today may be unaware of the details within each bill as currently written.
That existing regulatory arrangement will change if one of the two pending bills in Congress were to pass and be signed into law. That proposal is known as the Verifying Accurate Leading-Edge IVCT Development Act, or VALID Act. It is a bipartisan, 245-page bill that proposes FDA oversight of LDTs and is making its way through both the Senate and the House of Representatives.
A smaller, seven-page counterproposal is also before the Senate called the Verified Innovative Testing in American Laboratories Act, or VITAL Act. The VITAL Act would keep LDTs under CLIA but mandate updates to CLIA’s rules to account for modern tests.
Readers: Are you in favor of more or less regulation of LDTs? Take this quick survey and let us know what you think. Dark Daily wants to know your thoughts about LDT oversight. Click here to take our six-question survey. Results of this survey will be reported in a coming Dark Daily e-briefing.
Alert pathologists and clinical laboratory managers know that behind every bill proposed in Congress is a party with a vested interest that brought the issue to a senator or representative. Once enacted into law, a new bill changes the status quo, generally to the benefit of the private interests that requested that bill. This is true of both the VALID Act and the VITAL Act.
The table at the bottom of this briefing compares the provisions of each act and is current as of March 28.
Who Opposes VALID Act?
The VALID Act is garnering more attention than the VITAL Act.
On March 22, the American Association for Clinical Chemistry (AACC) sent out an email message urging its members to oppose the VALID Act.
“Let your legislators know that that if VALID becomes law, your institution and other hospitals and small commercial laboratories could be forced to stop providing LDTs,” wrote Patricia Jones, PhD, DABCC, FACB, Chair of AACC’s Policy and External Affairs Core Committee. The AACC has long criticized the VALID Act..
On the other side of the debate, Philadelphia-based The Pew Charitable Trusts, a nonprofit that in part analyzes publics policy, has come out in support of the VALID Act’s proposed requirements.
Two bills are pending in Congress about the future of LDT regulation.
“Although the [current] LDT regulatory process offers labs significant flexibility and enables a more rapid response to public health needs when no FDA-cleared or -approved test exists, the relative lack of oversight for LDTs puts the health of patients at risk,” Pew wrote in an October 2021 report on LDTs.
The Advanced Medical Technology Association also supports the VALID Act, as do many manufacturers of in vitro test kits and large commercial labs. Proponents also believe FDA regulation is needed for IVCTs because they are similar to medical devices and bring with them patient safety concerns.
The American Clinical Laboratory Association and the National Independent Laboratory Association (NILA) have not taken formal positions on the VALID Act.
Congress Could Roll VALID Act into MDUFA Vote to Win Passage
There may be an effort to attach the VALID Act to the authorization vote for the Medical Device User Fee Agreement V (MDUFA), according to a February health legislation alert from law firm Akin Gump Strauss Hauer & Feld based in Washington.
MDUFA funding provides resources to the FDA’s medical device review program. Congress is set to receive final MDUFA V recommendations in April.
Nineteen healthcare and lab industry groups, including the American Medical Association, AACC, AMP, and NILA, sent a joint letter to four Congress members on Feb. 23 requesting they deliberate the VALID Act separately and not as part of MDUFA.
Again, please complete this survey and tell us what you think about FDA regulation of LDTs, as defined in the VALID Act, compared to continuing LDT oversight via a modernized CLIA in the VITAL Act.
—Scott Wallask
Comparison of VALID Act and VITAL Act
VALID Act
VITAL Act
Full act name
Verifying Accurate Leading-Edge IVCT Development Act
Verified Innovative Testing in American Laboratories Act
Bill numbers
House Bill H.R.4128 Senate Bill S.2209
Senate Bill S.1666
Sponsors
Sen. Michael Bennet (D-CO) , Sen. Mike Braun (R-IN), Rep. Larry Bucshon, MD (R-IN), Sen. Richard Burr (R-NC), and Rep. Diana DeGette (D-CO)
Sen. Rand Paul (R-KY)
Provisions
Developers shall apply for premarket approval of IVCTs if there is insufficient evidence of analytical validity or clinical validity or if it’s reasonably possible an IVCT will cause serious adverse health effects.
Applications shall include a summary of test data and scientific evidence to support analytical and clinical validity of the test.
Through a technology certification, developers can submit an IVCT to the FDA for review, and if granted, the certification allows them to develop similar tests without going back for review each time.
The FDA must establish a program for rapid review of breakthrough IVCTs that provide effective treatment of life-threatening diseases
The federal government should work to ensure that regulatory oversight of laboratory tests does not limit patient access, impede innovation, or limit a test’s sustainability as a result of being unduly burdensome or beyond the fiscal capacity of the laboratory to reasonably validate and perform.
No aspects of LTDs shall be regulated under the FDA.
No later than 180 days after enactment of the bill, the secretary of health and human services shall report to the Senate’s Committee on Health, Education, Labor, and Pensions about recommendations to update clinical lab regulations and provide an assessment of LDT use during the 2020 pandemic response.
Exemptions
IVCTs being marketed before the VALID Act goes into effect
Low-risk tests
IVCTs that are granted emergency use
No new exemptions
Review timelines
The FDA shall make a decision no later than 90 days after an application is submitted.
No new requirements noted.
Sources: VALID Act and VITAL Act bills. Information is current as of March 28, 2022.
Proof of vaccination, masking, and availability of on-site testing will continue to be measures taken at in-person events for pathologists and medical laboratory professionals
Organizers of in-person clinical laboratory conferences face an interesting dilemma as they plan events in 2022: Where do they draw the line with COVID-19 safety protocols?
On one hand, the surge of cases caused by the SARS-CoV-2 Omicron variant seems to be in its waning stages and large swaths of the population are vaccinated. On the other hand, clinical laboratory and anatomic pathology events want potential registrants to have confidence that it is safe to travel and attend the gatherings.
One lab industry conference producer who happens to be knee-deep in preparing for an in-person meeting this spring is Robert Michel, Editor-in-Chief of The Dark Report and Founder of the 27th Annual Executive War College on Laboratory and Pathology Management. This informative event takes place on April 27-28 in New Orleans and includes COVID-19 protocols to protect attendees.
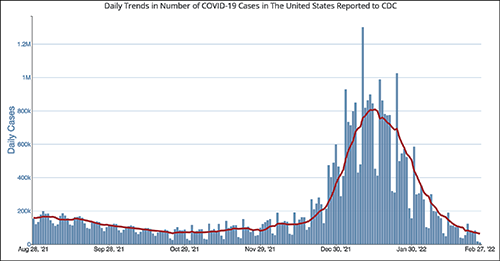
The CDC chart above shows the daily number of new COVID-19 cases in the US for the six-month period ending Feb. 28, 2022. Clinical laboratory managers should note that the number of new cases is at its lowest level since the Omicron variant showed up early this year.
“It’s important for all those planning to attend this year’s Executive War College to know that screening COVID-19 protocols will be in place to ensure the health and safety of all participants,” Michel noted. “We did a large lab conference in the fall of 2021 that included protocols for COVID-19 and the attendees told us they appreciated the protection provided by those protocols.”
After a significant rise in COVID-19 cases in January 2022 due to the Omicron variant, current daily case levels now are lower than they were six months ago before the new variant hit, according to numbers from the federal Centers for Disease Control and Prevention (CDC).
The in-person 2021 Executive War College, which took place in San Antonio on Nov. 2-3, 2021, followed the CDC’s recommendations:
COVID-19 protocols included a daily set of questions and a temperature check for all speakers and attendees before they were allowed to enter the conference area.
CLIA-complex rapid PCR COVID-19 tests were available for individuals whose temperature and answers to the screening questions indicated the need for such testing.
Attendees used an app to answer the daily screening questions and upload proof of vaccination.
“At last fall’s Executive War College, approximately 400 attendees were screened on each of the three days before entering the conference area and not one rapid COVID-19 test was needed,” Michel said. “Not only is that an outstanding outcome, but a number of attendees also told us they appreciated our efforts to keep them safe and protect their health.”
The 2022 Executive War College will follow the CDC’s updated COVID-19 guidelines, along with any state and local directives in effect as of April 27.
Although 300 attendees were expected at the 2021 Executive War College, 400 registered and participated.
Proof of Vaccination Has Been Required at Other Clinical Lab Industry Events
Organizers of other clinical lab conferences also have dealt with COVID-19 safety protocols. For example, the American Clinical Laboratory Association (ACLA) will hold its annual meeting in Washington, D.C., on March 9. COVID-19-related requirements for attendees will include proof of vaccination uploaded to a vaccine verification vendor and proof of a negative PCR test taken within 72 hours prior to the event.
The annual meeting of the American Society of Clinical Pathology (ASCP) occurs later this year in September in Chicago—too early yet to publish protocols. Last year’s ASCP conference in Boston was a hybrid event, offering both in-person and virtual options. Those who attended in person needed to upload proof of vaccination to a third-party vendor and were required to wear masks. On-site COVID-19 testing was available.
Revived Corporate Travel Could Boost Clinical Laboratory Conferences
The path back to live events across all industries has not been easy given various COVID-19 surges, political divisiveness over masking, frozen corporate travel budgets, and corporate policies banning or limiting employee travel.
Conference organizers throughout the United States universally hope those barriers will lower as 2022 progresses.
“With the fast-spreading Omicron triggering another round of setbacks to start 2022, event planners now are betting on spring to finally mark a turning point for the hard-hit industry,” MarketWatch reported on Feb. 4. “Their hopes hinge on American corporations taking a note from the recovery already under way for domestic air travel for leisure purposes, with the linchpin being a robust revival of trade show attendance and other in-person business gatherings.”
For Michel, offering actionable advice through well-thought-out sessions has been a cornerstone of the content offered each year at the Executive War College. He believes that approach will continue to be the strongest drawing point for clinical laboratory and pathology executives now considering attending the event.
“Our reading of the tea leaves is that across the profession of laboratory medicine, a great many managers, administrators, executives, and pathologists want to return to in-person conferences,” Michel noted. “Registrations for our April event are running ahead of 2019, and people tell us that they recognize the changes in healthcare and the lab marketplace because of the pandemic. They want to understand what’s driving current trends, like greater consumer involvement in lab testing and how to get private payers to reimburse claims for COVID-19 and genetic tests, as well as how a growing number of clinical laboratories are incorporating artificial intelligence solutions in both clinical care settings and lab operations.”
Pooled testing could become a critical tool for clinical laboratories to spot the SARS-CoV-2 coronavirus among asymptomatic and pre-symptomatic individuals
COVID-19 testing for individuals has expanded in the US, but the number of people actually tested remains a small proportion of the country’s total population and clinical laboratory testing supply shortages continue to hamper progress. A technique known as pooled testing may help. Federal experts hope it will substantially increase the number of individuals who are tested for the SARS-CoV-2 coronavirus before it makes a possible resurgence in the fall.
One-by-one, some of the nation’s largest clinical laboratory organizations are developing the capability to do pooled testing. For example, on July 18, the Food and Drug Administration (FDA) announced it had issued Quest Diagnostics (NYSE:DGX) an Emergency Use Authorization (EUA) for its SARS-CoV-2 rRT-PCR test, and that it is valid for up to four individual samples as a pooled test.
Quest’s rRT-PCR test was the first COVID-19 diagnostic test to be authorized for use with pooled samples, the FDA noted in a new release.
In the FDA’s statement announcing Quest’s EUA for its rRT-PCR test, Stephen M. Hahn, MD (above), FDA Commissioner, said, “This EUA for sample pooling is an important step forward in getting more COVID-19 tests to more Americans more quickly while preserving testing supplies.” He added, “Sample pooling becomes especially important as infection rates decline and we begin testing larger portions of the population.” (Photo copyright: CBS News.)
Following the announcement of Quest’s EUA, on July 24 the FDA announced LabCorp’s (NYSE:LH) EUA for its COVID-19 real-time reverse transcription polymerase chain reaction (rRT-PCR) test. The test, the EUA states, is intended for the “qualitative detection of nucleic acid from SARS-CoV-2 in upper and lower respiratory specimens” in individuals suspected of COVID-19, using “a matrix pooling strategy (i.e., group pooling strategy), containing up to five individual upper respiratory swab specimens (nasopharyngeal, mid-turbinate, anterior nares or oropharyngeal swabs) per pool and 25 specimens per matrix.”
Exponentially Increasing Testing
In pooled testing, instead of performing a coronavirus test on every specimen received by a clinical laboratory, samples from each individual specimen are taken and then combined with samples from other specimens. A single test is then performed on the entire collection of specimen samples.
If the results of the pooled samples are negative for coronavirus, it is safe to assume that all the specimens in the batch are negative for the virus. If the pooled sample comes back positive, then it will be necessary to go back to the original specimens in that pooled sample and test each specimen individually.
In an exclusive interview with Dark Daily’s sister print publication The Dark Report, Steven H. Hinrichs, MD, Chair of the Department of Pathology and Microbiology at the University of Nebraska Medical Center (UNMC), noted that one pitfall of pooled testing is that it works best in areas of low virus prevalence.
“For pooled testing, the ideal level of low prevalence would be an infection rate below 10%,” he said, adding, “For COVID-19 test manufacturers, pooled testing has the potential to reduce the number of standard tests labs run by roughly 40% to 60%, depending on the population being tested.
“Cutting the number of COVID-19 tests would be a disadvantage for test manufacturers, because pooled tests would identify large numbers of uninfected individuals who would not require standard testing with EUA tests.
“On the other hand, this policy would be a significant advantage for US labs because pooled testing would cut the number of standard tests,” he continued. “Clinical labs would save money on tests, reagents, and other supplies. It would also ease the burden on the lab’s technical staff,” Hinrichs concluded.
“In our study, we show that it’s reasonable to pool five samples, although we realized that some people may want to pool 10 samples at once,” noted Hinrichs. “But even if one sample is positive in a pool of five, then testing five samples at once saves 80% of our costs if all of those samples are negative. But, if one sample is positive, each of those five samples needs to be retested using the standard test,” Hinrichs explained.
During an American Society for Microbiology (ASM) virtual conference, Deborah Birx, MD, White House Coronavirus Response Coordinator, said, “Pooling would give us the capacity to go from a half a million tests per day to potentially five million individuals tested per day,” STAT reported.
Advantages of using pooled testing for the coronavirus include:
Expanding the number of individuals tested,
Stretching laboratory supplies, and
Reducing the costs associated with testing.
Health officials believe that individuals who have COVID-19 and are asymptomatic are largely responsible for the rising number of coronavirus cases in the US, STAT reported.
“It allows you to test more frequently in a population that may have a low prevalence of disease,” Benjamin Pinsky, MD, PhD, Associate Professor, Departments of Pathology and Medicine at Stanford University School of Medicine, told STAT. “That would allow you to test a lot of negatives, but also identify individuals who are then infected, before they develop symptoms.”
Pooled testing also could be advantageous for communities where COVID-19 is not prevalent, in neighborhoods that need to be tested during an outbreak, and for schools, universities, organizations, and businesses that want to remain safely open while periodically monitoring individuals for the virus, CNN reported.
“The goal is to increase the capacity of testing in a relatively straightforward fashion,” Pinsky told STAT. “The caveat is that by pooling the sample, you’re going to reduce the sensitivity of the test.”
According to Pinsky, “pooling only makes sense in places with low rates of COVID-19, where you expect the large majority of tests to be negative. Otherwise, too many of the pools would come back positive for it to work as a useful surveillance tool,” STAT reported.
As Clinical Lab Testing Increases, Pooled Testing for COVID-19 Could Be Critical
Pooled testing has been used in other countries, including China, to test larger amounts of people for COVID-19.
“If you look around the globe, the way people are doing a million tests or 10 million tests is they’re doing pooling,” Birx said during the ASM virtual conference, CNN reported.
In a press release, the American Clinical Laboratory Association (ACLA) stated that about 300,000 tests for COVID-19 were performed per day in labs across the US in late June. That number was up from approximately 100,000 tests being performed daily in early April.
“All across the country, clinical laboratories are increasing the number of labs processing tests, purchasing additional testing platforms, and expanding the number of suppliers to provide critical testing materials,” said Julie Khani, ACLA President in the press release. “However, the reality of this ongoing global pandemic is that testing supplies are limited. Every country across the globe is in need of essential testing supplies, like pipettes and reagents, and that demand is likely to increase in the coming months.”
Clinical laboratory managers will want to keep an eye on these developments. As the need for COVID-19 testing increases, pooled testing may provide an efficient, cost-effective way to spot the coronavirus, especially among those who are asymptomatic or pre-symptomatic and who display no symptoms.
Pooled testing could become a critical tool in the diagnosis of COVID-19 and potentially decrease the overall number of deaths.