The St. Louis-based in vitro diagnostics (IVD) developer is making PrecivityAD available to physicians while awaiting FDA clearance for the non-invasive test
Clinical laboratories have long awaited a test for Alzheimer’s disease and the wait may soon be over. The first blood test to aid physicians and clinical laboratories in the diagnosis of patients with memory and cognitive issues has been released by C₂N Diagnostics of St. Louis. The test measures biomarkers associated with amyloid plaques in the brain—the pathological hallmark of Alzheimer’s.
In a news release, PrecivityAD describes the laboratory-developed test (LDT) as “a highly sensitive blood test using mass spectrometry and is performed in C₂N’s CLIA-certified laboratory. While the test by itself cannot diagnose Alzheimer’s disease … the test is an important new tool for physicians to aid in the evaluation process.”
PrecivityAD provides physicians with an Amyloid Probability Score (APS) for each patient. For example:
A low APS (0-36) is consistent with a negative amyloid PET scan result and, thus, has a low likelihood of amyloid plaques, an indication other causes of cognitive symptoms should be investigated.
An intermediate APS (37-57) does not distinguish between the presence or absence of amyloid plaques and indicates further diagnostic evaluation may be needed to assess the underlying cause(s) for the patient’s cognitive symptoms.
A high APS (58-100) is consistent with a positive amyloid positron-emission tomography (PET) scan result and, thus, a high likelihood of amyloid plaques. Presence of amyloid plaques is consistent with an Alzheimer’s disease diagnosis in someone who has cognitive decline, but alone is insufficient for a final diagnosis.
The $1,250 test is not currently covered by health insurance or Medicare. However, C₂N Diagnostics has pledged to offer discounts to patients based on income levels.
Jeff Cummings, MD, ScD (above) Research Professor, Department of Brain Health, University of Nevada, Las Vegas, said in a C₂N Diagnostics press release, “A blood test for Alzheimer’s is a game changer.” While there is no cure for Alzheimer’s, a non-invasive blood test can help providers diagnose patients when their symptoms are mild and often misdiagnosed. “Advances in Alzheimer’s diagnostics are key to more effective identification, diagnosis, and clinical trial recruitment,” he added. Currently, brain changes caused by the disease are most commonly identified through PET scans. (Photo copyright: University of Nevada Las Vegas.)
Additional Research Requested
While C₂N’s PrecivityAD is the first test of its kind to reach the commercial market, it has not received US Food and Drug Administration (FDA) clearance, nor has the company published detailed data on the test’s accuracy. However, the PrecivityAD website says the laboratory-developed test “correctly identified brain amyloid plaque status (as determined by quantitative PET scans) in 86%” of 686 patients, all of whom were older than 60 years of age with subjective cognitive impairment or dementia.
But some Alzheimer’s advocacy groups are tempering their enthusiasm about the breakthrough. Eliezer Masliah, MD, Director of the Division of Neuroscience, National Institute on Aging, told the Associated Press (AP), “I would be cautious about interpreting any of these things,” he said of the company’s claims. “We’re encouraged, we’re interested, we’re funding this work, but we want to see results.”
Heather Snyder, PhD, Vice President, Medical and Scientific Relations at the Alzheimer’s Association told the AP her organization will not endorse a test without FDA clearance. The Alzheimer’s Association also would like to see the test studied in larger and diverse populations. “It’s not quite clear how accurate or generalizable the results are,” she said.
Braunstein defended the decision to make the test for Alzheimer’s immediately available to physicians, asking in the AP article, “Should we be holding that technology back when it could have a big impact on patient care?”
Howard Fillit, MD, Founding Executive Director and Chief Science Officer of the Alzheimer’s Drug Discovery Foundation (ADDF), maintains the first-of-its-kind blood test is an important milestone in Alzheimer’s research. ADDF invested in C₂N’s development of the test.
“Investing in biomarker research has been a core goal for the ADDF because having reliable, accessible, and affordable biomarkers for Alzheimer’s diagnosis is step one in finding drugs to prevent, slow, and even cure the disease,” Fillit said in an ADDF news release.
C₂N is also developing a Brain Health Panel to detect multiple blood-based markers for Alzheimer’s disease that will aid in better disease staging, treatment monitoring, and differential diagnosis.
Second Alzheimer’s Test in Development
Soon medical laboratories may have two different in vitro diagnostic tests for Alzheimer’s disease. On December 2, Fujirebio Diagnostics filed for FDA 510(k) premarket clearance for its Lumipulse G β-Amyloid Ratio (1-42/1-40) test, which looks for biomarkers found in cerebral spinal fluid.
“Accurate and earlier intervention will also facilitate the development of new drug therapies, which are urgently needed as the prevalence of Alzheimer’s disease increases with a rapidly aging population globally,” Fujirebio Diagnostics President and CEO Monte Wiltse said in a news release.
The Lumipulse G β-Amyloid test, which is intended for use in patients aged 50 and over presenting with cognitive impairment, has received CE-marking for use in the European Union.
Clinical laboratory managers will want to keep a close eye on rapidly evolving developments in testing for Alzheimer’s disease. It is the sixth leading cause of death in the United States and any clinical laboratory test that could produce an early and accurate diagnosis of Alzheimer’s Disease would become a valuable tool for physicians who treat patients with the symptoms of Alzheimer’s.
The AI protein-structure-prediction system may ‘revolutionize life sciences by enabling researchers to better understand disease,’ researchers say
Genomics leaders watched with enthusiasm as artificial intelligence (AI) accelerated discoveries that led to new clinical laboratory diagnostic tests and advanced the evolution of personalized medicine. Now Google’s London-based DeepMind has taken that a quantum step further by demonstrating its AI can predict the shape of proteins to within the width of one atom and model three-dimensional (3D) structures of proteins that scientist have been trying to map accurately for 50 years.
Pathologists and clinical laboratory professionals know that it is estimated that there are around 30,000 human genes. But the human proteome has a much larger number of unique proteins. The total number is still uncertain because scientists continue to identify new human proteins. For this reason, more knowledge of the human protein is expected to trigger an expanding number of new assays that can be used by medical laboratories for diagnostic, therapeutic, and patient-monitoring purposes.
DeepMind’s AI tool is called AlphaFold and the protein-structure-prediction system will enable scientists to quickly move from knowing a protein’s DNA sequence to determining its 3D shape without time-consuming experimentation. It “is expected to accelerate research into a host of illnesses, including COVID-19,” BBC News reported.
This protein-folding breakthrough not only answers one of biology’s biggest mysteries, but also has the potential to revolutionize life sciences by enabling researchers to better understand disease processes and design personalized therapies that target specific proteins.
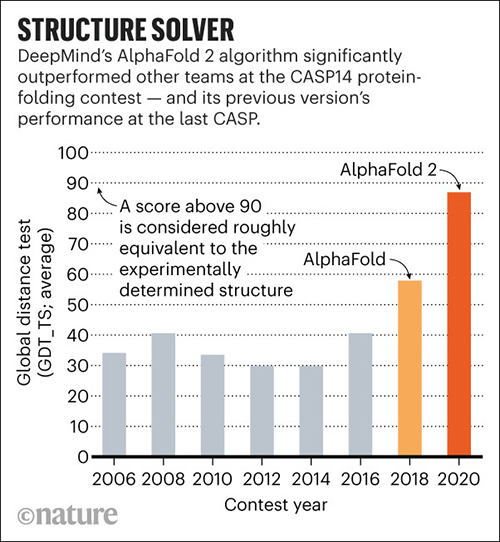
In November, DeepMind’s AlphaFold won the 14th Community Wide Experiment on Critical Assessment of Techniques for Protein Structure Prediction (CASP14), a biennial competition in which entrants receive amino acid sequences for about 100 proteins whose 3D structures are unknown. By comparing the computational predictions with the lab results, each CASP14 competitor received a global distance test (GDT) score. Scores above 90 out of 100 are considered equal to experimental methods. AlphaFold produced models for about two-thirds of the CASP14 target proteins with GDT scores above 90, a CASP14 press release states.
According to MIT Technology Review, DeepMind’s discovery is significant. That’s because its speed at predicting the structure of proteins is unprecedented and it matched the accuracy of several techniques used in clinical laboratories, including:
Unlike the laboratory techniques, which, MIT noted, are “expensive and slow” and “can take hundreds of thousands of dollars and years of trial and error for each protein,” AlphaFold can predict a protein’s shape in a few days.
“AlphaFold is a once in a generation advance, predicting protein structures with incredible speed and precision,” Arthur D. Levinson, PhD, Founder and CEO of Calico Life Sciences, said in a DeepMind blogpost. “This leap forward demonstrates how computational methods are poised to transform research in biology and hold much promise for accelerating the drug discovery process.”
Science reported that AlphaFold, which scored a median of 87—25 points above the next best predictions—did so well that CASP14 organizers worried DeepMind may have been somehow cheated. To validate the results, they asked AlphaFold to complete a “special challenge”—modeling a membrane protein from an ancient species of microbes called archaea, which they had been unable to model satisfactorily using X-ray crystallography. AlphaFold returned a detailed image of a three-part protein with two long helical arms in the middle. “It’s almost perfect,” Andrei Lupas, PhD, Director at the Max Planck Institute for Developmental Biology, told Science. “They could not possibly have cheated on this. I don’t know how they do it.” (Graphic copyright: DeepMind/Nature.)
“Even tiny rearrangements of these vital molecules can have catastrophic effects on our health, so one of the most efficient ways to understand disease and find new treatments is to study the proteins involved,” Moult said in the CASP14 press release. “There are tens of thousands of human proteins and many billions in other species, including bacteria and viruses, but working out the shape of just one requires expensive equipment and can take years.”
Science reported that the 3D structures of only 170,000 proteins have been solved, leaving roughly 200 million proteins that have yet to be modeled. Therefore, AlphaFold will help researchers in the fields of genomics, microbiomics, proteomics, and other omics understand the structure of protein complexes.
“Being able to investigate the shape of proteins quickly and accurately has the potential to revolutionize life sciences,” Andriy Kryshtafovych, PhD, Project Scientist at University of California, Davis, Genome Center, said in the press release. “Now that the problem has been largely solved for single proteins, the way is open for development of new methods for determining the shape of protein complexes—collections of proteins that work together to form much of the machinery of life, and for other applications.”
Clinical laboratories play a major role in the study of human biology. This breakthrough in genomics research and new insights into proteomics may provide opportunities for medical labs to develop new diagnostic tools and assays that better identify proteins of interest for diagnostic and therapeutic purposes.
Washington Post investigation outlines scientists’ frustrations in the early days of the pandemic, as they worked to deploy laboratory-developed tests for the novel coronavirus
In the wake of the failed rollout of the Centers for Disease Control and Prevention’s (CDC) COVID-19 diagnostic test last February, many CLIA-certified academic and public health laboratories were ready, and had the necessary resources, to develop their own coronavirus molecular diagnostic tests to help meet the nationwide demand for clinical laboratory testing. However, the response from the US Food and Drug Administration (FDA) was, in essence, “not so fast.”
In this second part of Dark Daily’s two-part e-briefing, we continue our coverage of the Washington Post (WP) investigation that detailed the regulatory hurdles which blocked private laboratories from deploying their own laboratory-developed tests (LDTs) for COVID-19. The report is based on previously unreported email messages and other documents reviewed by the WP, as well as the newspaper’s exclusive interviews with scientists and officials involved.
The CDC’s COVID-19 test kits began arriving at public health laboratories on February 8, just 18 days after the first case of the novel coronavirus was confirmed in the US. As the WP noted in an earlier analysis, titled, “What Went Wrong with Coronavirus Testing in the US,” the CDC’s decision to develop its own test was not surprising. “The CDC will develop [its] own test that is suited to an American healthcare context and the regulations that exist here,” explained Jeremy Konyndyk, Senior Policy Fellow at the Center for Global Development. “That’s how we normally would do things.”
But state and local public health laboratories quickly discovered that the CDC test kits were flawed due to problems with one of the reagents. While numerous academic, research, and commercial labs had the capability to produce their own COVID-19 PCR tests, FDA rules initially prevented them from doing so without a federal Emergency Use Authorization (EUA).
The bureaucratic hurdles arose due to Health and Human Services Secretary Alex Azar’s January 31 declaration that COVID-19 was a “health emergency” in the US. By doing so, HHS triggered a mandate that requires CLIA-certified labs at universities, research centers, and hospitals to seek an EUA from the FDA before deploying any laboratory-developed tests.
Scientists, Clinical Laboratories Frustrated by Bureaucratic Delays and Red Tape
To make matters worse, the EUA process was neither simple nor fast, which exasperated lab scientists and clinical laboratory administrators. “In their private communications, scientists at academic, hospital, and public health labs—one layer removed from federal agency operations—expressed dismay at the failure to move more quickly, and frustration at bureaucratic demands that delayed their attempts to develop alternatives to the CDC test,” wrote the WP investigators.
In a Feb. 27 email to other microbiologists, Marc Couturier, PhD, Medical Director at ARUP Laboratories, a national reference laboratory network located in Utah, voiced his irritation with the red tape that stymied private laboratory development of COVID-19 tests. He wrote, “We have the skills and resources as a community, but we are collectively paralyzed by a bloated bureaucratic/administrative process,” reported the WP.
Keith Jerome, MD, PhD (above), Head of the Virology Division at the Fred Hutchinson Cancer Research Center in Seattle, maintains federal regulations muted one of the nation’s greatest assets in the fight against COVID-19. “The great strength the US has always had, not just in virology, is that we’ve always had a wide variety of people and groups working on any given problem,” he told MIT Technology Review. “When we decided all coronavirus testing had to be done by a single entity, even one as outstanding as CDC, we basically gave away our greatest strength.” (Photo copyright: Jonathan Hamilton/NPR.)
‘FDA Should Not Treat Labs Like They Are Creating Commercial Products’
According to Kaiser Health News (KHN), Greninger was able to identify one of the nation’s first cases of community-acquired COVID-19 by taking “advantage of a regulatory loophole that allowed the lab to test samples obtained for research purposes from UW’s hospitals.”
But navigating the EUA process was a different story, Greninger told the WP. He spent more than 100 hours filling out forms and collecting information needed for the EUA application. After emailing the application to the FDA, Greninger received a reply containing eCopy Guidance telling him he needed to resubmit the information to the Document Control Center (DCC) at the Center for Devices and Radiological Health (CDRH), a federal agency Greninger knew nothing about. Another FDA rule required that the submission be copied to a hard disk and mailed to the DCC.
In an interview with ProPublica, Greninger stated that after he submitted his COVID-19 test—which copies the CDC protocol—an FDA reviewer told him he would need to prove the test would not show a positive result for someone infected with either a SARS or MERS coronavirus. The first SARS coronavirus disappeared in mid-2003 and the only two cases of MERS in the US were diagnosed in 2014. Greninger told ProPublica it took him two days to locate a clinical laboratory that could provide the materials he needed.
Greninger maintains the FDA should not treat all clinical laboratories as though they are making a commercial product. “I think it makes sense to have this regulation when you’re going to sell 100,000 widgets across the US. That’s not who we are,” he told ProPublica.
FDA Changes Course
Under pressure from clinical laboratory scientists and medical doctors, by the end of February the FDA had issued new policy that enabled CLIA-certified laboratories to immediately use their validated COVID-19 diagnostics while awaiting an EUA. “This policy change was an unprecedented action to expand access to testing,” said the FDA in a statement.
Since then, the FDA has continued to respond—albeit slowly—to scientists’ complaints about regulations that hampered the nation’s COVID-19 testing capacity.
Clinical laboratory leaders and pathologists involved in testing for the SARS-CoV-2 coronavirus should monitor the FDA’s actions and be aware of when and if certain temporary changes the agency implemented during the early days of the COVID-19 pandemic become permanent.
To read part one of our two-part coverage of the Washington Post’s investigation, click here.
Previously unreported email messages and documents paint vivid picture of public health laboratory officials’ dismay and frustration over testing delays
Between late January and early March, Clinical laboratory leaders watched with dismay as federal government missteps crippled the Centers for Disease Control and Prevention’s (CDC) rollout of its COVID-19 diagnostic testing in the early days of the pandemic. The resulting lack of testing capacity enabled the novel coronavirus’ spread across the United States.
This first part of Dark Daily’s two-part e-briefing covers how investigators at the Washington Post (WP) have produced a timeline describing the CDC initial failure to produce a reliable laboratory test for COVID-19 and the regulatory hurdles that blocked medical laboratories from developing their own tests for the virus. The WP’s report is based on previously unreleased email messages and other documents reviewed by the WP, as well as the newspaper’s exclusive interviews with medical laboratory scientists and officials involved.
A New York Times report on the federal government’s initial review of the testing kit failure pinned the blame on sloppy practices at CDC laboratories in Atlanta and a lack of expertise in commercial manufacturing. However, the WP reported that COVID-19 testing kits were delayed due to a “glaring scientific breakdown” at the central lab, created when the CDC facilities that assembled the kits “violated sound manufacturing practices” that resulted in cross contamination of testing compounds.
The US and other countries have criticized China for a lack of transparency about the virus’ emergence, which came to light on December 31, 2019, when China reported a cluster of pneumonia cases in Wuhan, according to a World Health Organization (WHO) timeline. A week later, Chinese authorities identified the pneumonia-like illness as being caused by a new novel coronavirus.
In the US, the first case of COVID-19 was found January 21 in a Washington State man who had traveled to Wuhan. But in the weeks that followed, the US government’s inability to establish a systematic testing policy became the catalyst for the virus’ ultimate spread to more than two million people, notes the CDC website.
ProPublica, which conducted its own investigation into the early stages of the government’s coronavirus response, blamed the failures on “chaos” at the CDC and “an antiquated public health system trying to adapt on the fly.”
The CDC’s first mistake may have been underestimating the danger COVID-19 posed to public health in this country. During a January 15 conference call, CDC scientists assured state and county public health officials that the agency was developing a COVID-19 diagnostic test which soon would be available, but which may not be needed “unless the scope gets much larger than we anticipate right now,” reported the WP.
A week later, an interview with CNBC, President Trump said, “We have it under control. It’s going to be just fine.”
CDC scientists designed their test in seven days, which, according to the WP investigators, is “a stunningly short period of time for a healthcare system built around the principles of medical quality and patient safety, not speed.” But when those initial CDC-made tests arrived at a New York City public health laboratory on February 8, lab technicians discovered the COVID-19 assays often indicated the presence of the coronavirus in samples that the lab’s scientists knew did not contain the virus.
When the scientists informed Lab Director Jennifer Rakeman, PhD, Assistant Commissioner, New York City Department of Health and Mental Hygiene, her response, according to the WP, was “Oh, s—. What are we going to do now?”
That night, Director Jill Taylor, PhD, Director of New York State’s Wadsworth Center public health reference laboratory, emailed state health officials, stating, “There is a technical problem in one of the reagents which invalidates the assay and will not allow us to perform the assay,” reported the WP. “I’m sorry not to have better news.”
Scott Becker (above), Executive Director of the Association of Public Health Laboratories (APHL), voiced his concerns about the CDC’s flawed COVID-19 test kits in an email to a CDC official, reported the WP. “The states and their governors are going to come unglued,” Becker wrote, adding, “If the CDC doesn’t get ahead of this, it will be a disaster.” (Photo copyright: Bill O’Leary/The Washington Post.)
‘The Silence from CDC is Deafening’
On February 10, Joanne Bartkus, PhD, then-Lab Director of the Minnesota Health of Department, wrote to APHL Executive Director Scott Becker: “The silence from CDC … is deafening. What is going on?” reported the WP.
By the end of February, the Associated Press (AP) reported that only 472 patients had been tested for COVID-19 nationwide. By comparison, South Korea, which identified its first case of COVID-19 on the same day as the US, was testing 1,000 people per day.
A WHO spokesperson told the WP that, “… no discussions occurred between WHO and CDC (or other US government agencies) about WHO providing COVID-19 tests to the US.” When the CDC’s original COVID-19 test kit failed, there may not have been a Plan B. This may explain why the opportunity to contain COVID-19 through surveillance testing was lost during the weeks it took to design a fix for the CDC test and loosen regulations so clinical laboratories could develop their own tests.
As medical laboratory scientists and clinical laboratory leaders know, the lack of early COVID-19 testing was a public health failure and painted a false picture of the virus’ spread. Nearly five months after the first case of the virus was confirmed in the US, testing capacity may only now be outpacing demand.
Click here to read part two of our coverage of the Washington Post’s investigation.
This new technology could replace needle biopsies and allow physicians to detect rejection of transplanted organs earlier, saving patients’ lives
Anatomic pathologists
may be reading fewer biopsy reports for patients with organ transplants in the
future. That’s thanks to a new technology that may be more sensitive to and
capable of detecting organ rejection earlier than traditional needle biopsies.
When clinicians can detect organ transplant rejection
earlier, patients survive longer. Unfortunately, extensive organ damage may
have already occurred by the time rejection is detected through a traditional
needle biopsy. This led a group of researchers at Emory University School of Medicine to
search for a better method for detecting organ rejection in patients with transplants.
The Emory researchers describe the method and technology
they devised in a paper published in Nature Biomedical
Engineering, titled, “Non-Invasive Early Detection of Acute Transplant
Rejection Via Nanosensors of Granzyme B Activity.” The new technology could
make it easier for clinicians to detect when a patient’s body is rejecting a
transplanted organ at an earlier time than traditional methods.
This technology also provides a running measure of processes,
so clinicians have more powerful tools for deciding on the most appropriate
dosage of immunosuppressant
drugs.
“Right now, most tests are aimed at organ dysfunction, and
sometimes they don’t signal there is a problem until organ function is below 50
percent,” Andrew
Adams, MD, PhD Co-Principal Investigator and an Associate Professor of Surgery
at Emory University School of Medicine, in a Georgia
Institute of Technology news release.
How the Technology Works
The method that Adams and his colleagues tested involves the
detection of granzyme B,
a serine protease
often found in the granules of natural killer cells
(NK cells) and cytotoxic
T cells. “Before any organ damage can happen, T cells have to produce granzyme
B, which is why this is an early detection method,” said Gabe Kwong, PhD, Assistant
Professor in the Wallace H. Coulter Department of Biomedical Engineering at
Georgia Tech and Emory University, in the news release.
The new technology is made up of sensor nanoparticles in the
shape of a ball with iron oxide in the middle. Amino acids stick out of the
ball like bristles. Each amino acid has a fluorescent molecule attached to the
tip.
The nanoparticles are injected into the patient. Their size
prevents them from gathering in the patient’s tissue or from being flushed out
through the kidneys. They are designed to accumulate in the tissue of the
transplanted organ.
If the T cells in the transplanted organ begin to produce
granzyme B, the amino acids break away from the nanoparticles, releasing the
fluorescent molecules attached to their tips. Those molecules are small enough
to be processed through the kidneys and can be detected in the patient’s urine.
Pathologists Play Crucial Role on Transplant Teams
Anatomical pathologists (histopathologists in the UK) are key
members of transplant teams for many reasons, including their ability to assess
biopsies. The current method for detecting organ transplant rejection involves
needle biopsies. It is considered the gold standard.
However, according to a paper published in the International
Journal of Organ Transplantation Medicine: “Although imaging studies
and laboratory findings are important and helpful in monitoring of the
transplanted liver, in many circumstances they are not sensitive enough. For
conditions such as rejection of the transplant, liver histology remains the
gold-standard test for the diagnosis of allograft dysfunction. Therefore,
histopathologic assessments of allograft liver
biopsies have an important role in managing patients who have undergone liver
transplantation.”
There are two main problems with needle biopsies. The first,
as mentioned above, is that they don’t always catch the rejection soon enough.
The second is that the needle may cause damage to the transplanted organ.
“The biggest risk of a biopsy is bleeding and injury to the transplanted organ,” noted Andrew Adams, MD, PhD (above), Co-Principal Investigator and an Associate Professor of Surgery at Emory University School of Medicine, in the Georgia Tech news release. “Then there’s the possibility of infection. You’re also just taking a tiny fraction of the transplanted organ to determine what’s going on with the whole organ, and you may miss rejection or misdiagnose it because the needle didn’t hit the right spot,” he added.
And, according to Kwong, even though biopsies are the gold
standard, the results represent one moment in time. “The biopsy is not
predictive. It’s a static snapshot. It’s like looking at a photo of people in
mid-jump. You don’t know if they’re on their way up or on their way down. With
a biopsy, you don’t know whether rejection is progressing or regressing.”
Future Directions of Emory’s Research
The research conducted by Adams and Kwong, et al, is in its
early stages, and the new technology they created won’t be ready to be used on patients
for some time. Nevertheless, there’s reason to be excited.
Nanoparticles are not nearly as invasive as a needle biopsy.
Thus, risk of infection or damaging the transplanted organ is much lower. And Emory’s
technology would allow for much earlier detection, as well as giving clinicians
a better way to adjust the dose of immunosuppressant drugs the patient takes.
“Adjusting the dose is very difficult but very important
because heavy immunosuppression increases occurrence of infections and patients
who receive it also get cancer more often,” said Kwong. The new technology
provides a method of measuring biological activity rates, which would give
clinicians a clearer picture of what’s happening.
The Emory team’s plan is to enhance the new sensors to
detect at least one other major cause of transplant rejection—antibodies. When
a patient’s body rejects a transplanted organ, it produces antibodies to
neutralize what it sees as a foreign entity.
“Antibodies kill their target cells through similar types of
enzymes. In the future, we envision a single sensor to detect both types of
rejection,” said Kwong.
Adams adds, “This method could be adapted to tease out
multiple problems like rejection, infection, or injury to the transplanted
organ. The treatments for all of those are different, so we could select the
proper treatment or combination of treatments and also use the test to measure
how effective treatment is.”
This line of research at Emory University demonstrates how
expanding knowledge in a variety of fields can be combined in new ways. As this
happens, medical laboratories not only get new biomarkers that can be
clinically useful without the need for invasive procedures like needle biopsies,
but these same biomarkers can guide the selection of more effective therapies.