“The timing of EWC with the release of this policy couldn’t be better,” CEO and founder of Momentum ConsultingValerie Palmieri told Dark Daily in an interview at Monday night’s opening reception. “It’s a great conference to not only catch up with colleagues but really hear and have those difficult discussions about where we are today, where we’re going, and where we need to be.”
Final LDT rule ‘radically’ different than draft
Tim Stenzel, MD, PhD, former director of the FDA’s Office of In Vitro Diagnostics called the finalized rule “radically different” from the proposed rule. In some ways it is less complex: “The bar is lower,” he said, noting that he was voicing his personal views and not those of the federal agency. “I was convinced that there would be lawsuits, but I’m now not sure if that’s advisable.”
Still, laboratory teams will have to parse the more than 500-page document to determine how the final rule relates to their specific circumstances. After that, it won’t be as challenging, Stenzel said.
His advice: First, read the rule. Second, reach out to FDA for help—he’s sure, he said, that the office is geared up to respond to a “ton of questions” about the implications for individual labs and are standing by to answer emails from labs. And, he added in a discussion session, emailing the agency is free.
The final rule will be in force 60 days after it’s published. Stenzel provided a timeline for some of the milestones:
1 Year: Comply with MD(AE) reporting and reporting of corrections and removals.
2 Years: Comply with labeling, registration and listing, and investigational use requirements.
3 Years: QS records and, in some cases, design controls and purchasing controls.
3.5 Years: Comply with high risk (class III) premarket review requirements.
4 Years: Comply with moderate and low-risk premarket review requirements.
Executive Chair and CEO of XiFin, Inc.Lâle White welcomed the audience with a morning keynote entitled “Big Changes in Healthcare” on new regulations and diagnostics players poised to reshape lab testing.
The diagnostics business is in constant flux, she noted, from payer requirements to greater regulatory and compliance burdens on labs. Other factors include the growing senior population and increasingly complex health conditions, rising costs throughout the healthcare ecosystem, falling funding and reimbursement, and staffing shortages.
As for the economic challenges, consumers are increasingly making decisions based on cost, convenience and quality. The population is shifting to Medicare advantage, which is more cost effective. But changes to the star ratings system will mean lower pay for payer organizations. Those companies will, in turn, mitigate their losses by making changes to pre-authorizations and tightening denials, even for clean claims.
Still, White said, more money isn’t the answer.
White urged the audience to use technology, including artificial intelligence and advances in genetic testing, to manage these and other industry changes.
“We need to optimize the tests we order,” she said. “And if we did that, lab diagnostics really has the potential to change the economics of health and improve outcomes.”
The FDA, Stenzel added, is “very interested” in stimulating innovation, building on the laboratory industry’s success in responding swiftly to the COVID pandemic and outbreaks of Monkey Pox, for example.
He shared lessons learned from recent public health emergencies, talked about CDC’s efforts to engage with clinical labs to improve future public health readiness and response and provided an overview of the CDC’s first laboratory-specific center.
“Laboratories are fundamental to public health,” he said. The industry is on the “front lines” when it comes to identifying threats, responding to them, and preparing for future responses.
Robert Michel, Editor-in-Chief of The Dark Report wrapped up the day’s regulatory discussions with a general session on the “regulatory trifecta” that includes the LDT final rule, CLIA regulations, and private payers’ policies for genetic claims.
But even though the College of American Pathologists (CAP) and nine other organizations signed a December 12 stakeholder letter to leaders of key House and Senate committees urging passage of legislation that would enable some regulation of LDTs, the VALID Act was ultimately omitted from the year-end omnibus spending bill (H.R. 2617).
That may be due to pressure from organizations representing clinical laboratories and pathologists which lobbied hard against the bill.
Responding to criticism of its stance on FDA oversight of LDTs, in a May 2022 open letter posted on the organization’s website, anatomic pathologist and CAP president Emily Volk, MD, said “we at the CAP have an honest difference of opinion with some other respected laboratory organizations. … We believe the VALID Act is the only viable piece of legislation addressing the LDT issue. … the VALID Act contains many provisions that are similar to policy the CAP has advocated for regarding the regulation of laboratory tests since 2009. Importantly, the current version includes explicit protections for pathologists and our ability to practice medicine without infringement from the Food and Drug Administration (FDA).” (Photo copyright: College of American Pathologists.)
Organizations on Both Sides Brought Pressure to Bear on Legislators
The AAMC and AMP were especially influential, Bucshon told ProPublica. In addition to spending hefty sums on lobbying, AMP urged its members to contact legislators directly and provided talking points, ProPublica reported.
“The academic medical centers and big medical centers are in every state,” Bucshon said. As major employers in many locales, they have “a pretty big voice,” he added.
Discussing CAP’s reasoning behind its support of the VALID Act in a May 26 open letter and podcast, CAP president Emily Volk, MD, said the Valid Act “creates a risk-based system of oversight utilizing three tiers—low, moderate and high risk—in order to target the attention of the FDA oversight.”
While acknowledging that it had room for improvement, she lauded the bill’s three-tier risk-based system, in which tests deemed to have the greatest risks would receive the highest level of scrutiny.
She also noted that the bill exempts existing LDTs from an FDA premarket review “unless there is a safety concern for patients.” It would also exempt “low-volume tests, modified tests, manual interpretation tests, and humanitarian tests,” she wrote.
In addition, the bill would “direct the FDA not to create regulations that are duplicative of regulation under CLIA,” she noted, and “would require the FDA to conduct public hearings on LDT oversight.”
Pros and Cons of the VALID Act
One concern raised by opponents relates to how the VALID Act addressed user fees paid by clinical laboratories to fund FDA compliance activities. But Volk wrote that any specific fees “would need to be approved by Congress in a future FDA user fee authorization bill after years of public input.”
During the May 2022 podcast, Volk also cast CAP’s support as a matter of recognizing political realities.
“We understand that support for FDA oversight of laboratory-developed tests or IVCTs is present on both sides of the aisle and in both houses of Congress,” she said. “In fact, it enjoys wide support among very influential patient advocacy groups.” These groups “are very sophisticated in their understanding of the issues with laboratory-developed tests, and they do have the ear of Congress. There are many in the laboratory community that believe the VALID Act goes too far, but I can tell you that many of these patient groups don’t believe it goes far enough and are actively pushing for even more restrictive paradigms.”
Also urging passage of the bill were former FDA commissioners Scott Gottlieb, MD, and Mark B. McClellan, MD, PhD. In a Dec. 5 opinion piece for STAT, they noted that “diagnostic technologies have undergone considerable advances in recent decades, owing to innovation in fields like genomics, proteomics, and data science.” However, they wrote, laws governing FDA oversight “have not kept pace,” placing the agency in a position of regulating tests based on where they are made—in a medical laboratory or by a manufacturer—instead of their “distinctive complexity or potential risks.”
In their May 22 letter, opponents of the legislation outlined broad areas of concern. They contended that it would create “an onerous and complex system that would radically alter the way that laboratory testing is regulated to the detriment of patient care.” And even though existing tests would be largely exempted from oversight, “the utility of these tests would diminish over time as the VALID Act puts overly restrictive constraints on how they can be modified.”
CLIA Regulation of LDTs also Under Scrutiny
The provision to avoid duplication with the Clinical Laboratory Improvement Amendments (CLIA) program—which currently has some regulatory oversight of LDTs and IVCTs—is “insufficient,” opponents added, “especially when other aspects of the legislation call for requirements and activities that lead to duplicative and unnecessary regulatory burden.”
Opponents to the VALID Act also argued that the definitions of high-, medium-, and low-risk test categories lacked clarity, stating that “the newly created definition of moderate risk appears to overlap with the definition of high risk.”
The opponents also took issue with the degree of discretion that the bill grants to the US Secretary of Health and Human Services. This will create “an unpredictable regulatory process and ambiguities in the significance of the policy,” they wrote, while urging the Senate committee to “narrow the discretion so that stakeholders may better evaluate and understand the implications of this legislation.”
Decades ago, clinical laboratory researchers were allowed to develop assays in tandem with clinicians that were intended to provide accurate diagnoses, earlier detection of disease, and help guide selection of therapies. Since the 1990s, however, an industry of investor-funded laboratory companies have brought proprietary LDTs to the national market. Many recognize that this falls outside the government’s original intent for encouragement of laboratory-developed tests to begin with.
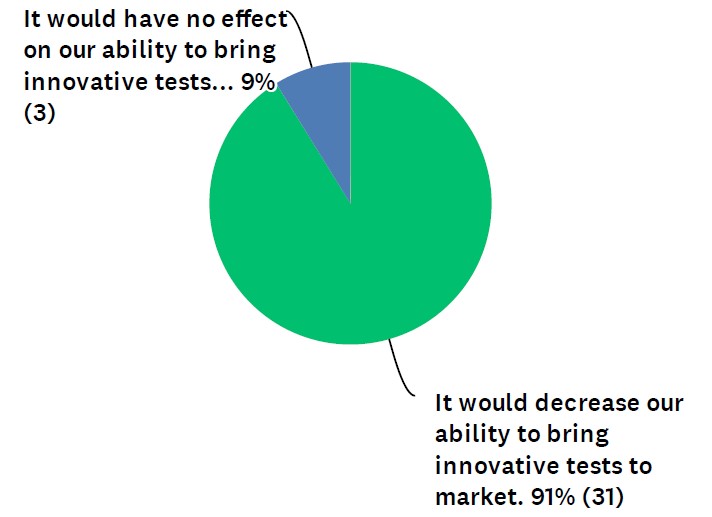
Though response was limited in Dark Daily’s poll, the message from respondents was overwhelmingly negative on LDT regulation by the FDA
Most respondents to a recent industry survey said that should Food and Drug Administration (FDA) approval be required in the future for laboratory developed tests (LDTs), innovation will suffer.
In the survey, which was conducted by Dark Daily, 91% of respondents said FDA pre-market approval of LDTs would decrease clinical laboratories’ ability to bring innovative tests to market. (See Figure 1.) The other 9% felt it would have no effect on innovation. Zero of the respondents said FDA involvement would increase innovative tests.
Figure 1: “How might a requirement of FDA pre-market approval impact the ability of your lab (or a lab you work for) to bring innovative tests to market?“
“Development of LDT tests has been the mission of most of the labs, and it meets the need for patient care,” noted one respondent in the survey. “Moving LDTs under FDA will create more obstacles for labs to offer the tests.”
To be fair, the survey had limited responses—34 in total. The poll went out to thousands of Dark Daily readers. We found that response rate surprising given how many labs will be affected if the VALID Act becomes law.
The VALID Act is a bipartisan bill that proposes FDA oversight of laboratory developed tests. The bill continues to make its way through the U.S. Senate and the House of Representatives.
A counterproposal called the Verified Innovative Testing in American Laboratories Act (VITAL Act) is also before Congress but has less momentum behind it. The VITAL Act seeks to keep LDTs under CLIA while also calling for reforms to account for modern lab tests.
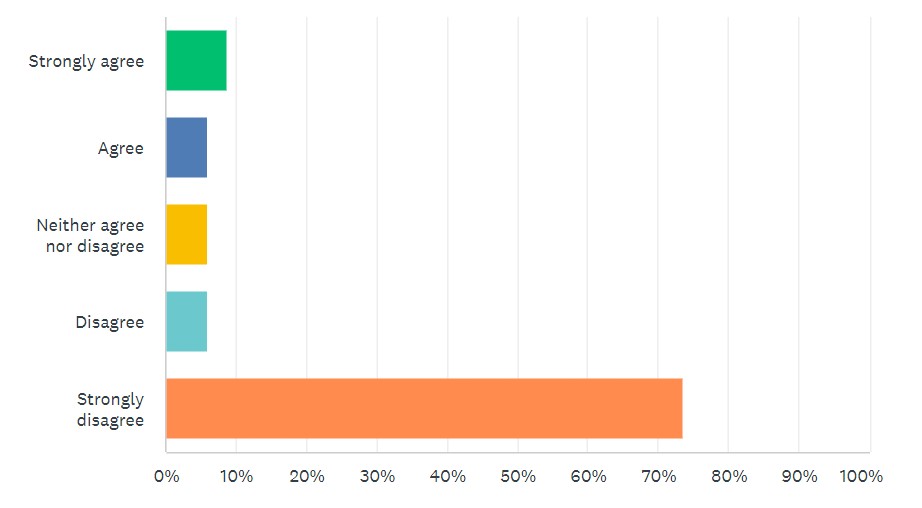
Looking at survey results, 80% “strongly disagreed” or “disagreed” that new LDT requirements, such as those found in the VALID Act, are needed. (See Figure 2.)
Figure 2: “FDA pre-market approval of LDTs should be required, as proposed in the VALID Act.”
By comparison, 65% “strongly agreed” or “agreed” with modernizing CLIA requirements for LDTs, as called for in the VITAL Act.
Those numbers shifted somewhat depending on the lab setting of the respondent. For example, just looking at commercial labs, opposition to the VALID Act remained similar, but support for modernizing CLIA jumped up to 88%. When looking at just hospitals, independent labs, and academic labs, numbers for both topics remained consistent.
When filtering the answers, the number of lab employees in a setting had little effect on survey results.
Political Battle Continues Over Laboratory Developed Tests
Clinical laboratory industry groups and others have been amassing to oppose or support the VALID Act. For example, the Advanced Medical Technology Association and The Pew Charitable Trusts are behind the bill.
However, the American Association for Clinical Chemistry, Association for Molecular Pathology, and new Coalition for Innovative Laboratory Testing are against the VALID Act.
Like Holmes, Balwani faces 12 counts of fraud and conspiracy to commit wire fraud for allegedly misleading investors, patients, and others about blood-testing startup’s technology
Clinical laboratory managers and pathologists are buckling up as the next installment of the Theranos story gets underway, this time for the criminal fraud trial of ex-Theranos President and COO Ramesh “Sunny” Balwani.
In one text to Holmes, Balwani wrote, “I am responsible for everything at Theranos,” NBC Bay Area reported.
Partners in Everything, including Crime, Prosecutors Allege
According to the Wall Street Journal (WSJ), prosecutors are following the Holmes trial playbook. They focused their opening arguments on the personal and working relationships between the pair, tying Balwani to Holmes’ crimes at the Silicon Valley blood-testing startup.
As second in command at Theranos, Balwani helped run the company from 2009 to 2016. He also invested $5 million in Theranos stock, while also underwriting a $13 million corporate loan.
“They were partners in everything, including their crimes,” Assistant US Attorney Robert Leach told jurors, the Mercury News reported. “The defendant and Holmes knew the rosy falsehoods that they were telling investors were contrary to the reality within Theranos.”
Leach maintained that Balwani was responsible for the phony financial projections Theranos gave investors in 2015 predicting $990 million in revenue when the company had less than $2 million in sales.
Former Theranos President and COO Ramesh “Sunny” Balwani (above) is seen arriving at the federal court in San Jose, California, for the start of his federal fraud trial. Clinical laboratory leaders and pathologists who followed the trial of ex-Theranos founder and CEO Elizabeth Holmes will no doubt be interested in what can be learned from this trail as well. (Photo copyright: Jim Wilson/The New York Times.)
“This is a case about fraud. About lying and cheating to obtain money and property,” Leach added. Balwani “did this to get money from investors, and he did this to get money and business from paying patients who were counting on Theranos to deliver accurate and reliable blood tests so that they could make important medical decisions,” the WSJ reported.
Defense attorneys downplayed Balwani’s decision-making role within Theranos, pointing out that he did not join the start-up until six years after Holmes founded the company with the goal of revolutionizing blood testing by developing a device capable of performing blood tests using a finger-prick of blood.
“Sunny Balwani did not start Theranos. He did not control Theranos. Elizabeth Holmes, not Sunny, founded Theranos and built Theranos,” defense attorney Stephen Cazares, JD of San Francisco-based Orrick, said in his opening argument, the WSJ reported.
The trial was expected to begin in January but was delayed by the unexpected length of the Holmes trial. It was then pushed out to March when COVID-19 Omicron cases spiked in California during the winter.
Balwani’s trial is being held in the same San Jose courthouse where Holmes was convicted. Balwani, 56, is facing identical charges as Holmes, which include two counts of conspiracy to commit wire fraud and 10 counts of wire fraud. He has pleaded not guilty.
Holmes, who is currently free on a $500,000 bond, will be sentenced on Sept. 26, Dark Daily reported in January.
Judge Excludes Jurors for Watching Hulu’s ‘The Dropout’
During jury selection in March, some jurors acknowledged they were familiar with the case, causing delays in impaneling the 12-member jury and six alternates. US District Court Judge Edward Davila excluded two potential jurors because they had watched “The Dropout,” Hulu’s miniseries about Holmes and Theranos. Multiple other jurors were dropped because they had followed the Holmes trial in the news, Law360 reported.
When testimony began, prosecutors had a familiar name take the stand—whistleblower and former Theranos lab tech Erika Cheung, who provided key testimony in the Holmes trial. During her testimony, Cheung said she revealed to authorities what she saw at Theranos because “Theranos had gone to extreme lengths to [cover up] what was happening in the lab,” KRON4 in San Francisco reported.
“It was important to report the truth,” she added. “I felt that despite the risk—and I knew there could be consequences—people really need to see the truth of what was happening behind closed doors.”
Nevada State Public Health Laboratory (NSPHL) Director Mark Pandori, PhD, who served as Theranos’ lab director from December 2013 to May 2014, was the prosecution’s second witness. Pandori testified that receiving accurate results for some tests run through Theranos’ Edison blood testing machine was like “flipping a coin.”
“When you are working in a place like Theranos, you’re developing something new. And you want it to work. Quality control remained a problem for the duration of my time at the company. There was never a solution to poor performance,” Pandori testified, according to KRON4.
While the defense team has downplayed Balwani’s decision-making role—calling him a “shareholder”—Aron Solomon, JD, a legal analyst with Esquire Digital, maintains they may have a hard time convincing the jury that Balwani wasn’t a key player.
“There’s no way the defense is going to be successful in painting Sunny Balwani in the light simply as a shareholder,” he told NBC Bay Area. “We know that, literally, Sunny Balwani was intimately involved with Theranos, because he was intimately involved with Elizabeth Holmes,” Solomon added.
Little Media Buzz for Balwani, Unlike Holmes Trial
While the Holmes trial hogged the media spotlight and drew daily onlookers outside the courthouse, reporters covering Balwani’s court appearances describe a much different atmosphere.
“The sparse crowd and quiet atmosphere at US District Court in San Jose, Calif., felt nothing like the circus frenzy that engulfed the same sidewalk months earlier when his alleged co-conspirator and former girlfriend, Elizabeth Holmes, stood trial on the same charges,” The New York Times noted in its coverage of the Balwani trial.
The Balwani trial may not reach the same headline-producing fervor as the Holmes legal battle. However, clinical laboratory directors and pathologists who follow these proceedings will no doubt come away with important insights into how Theranos went so terribly wrong and how lab directors must act under the Clinical Laboratory Improvement Amendments of 1988 (CLIA).
Medical laboratory employee alleges healthcare system discriminated based on her medical condition, failed to accommodate her disability, then retaliated and created hostile working conditions
What is a clinical laboratory’s obligation when an employee is infected with the SARS-CoV-2 coronavirus and does not make a speedy recovery? Medical laboratory executives should ponder this question now that a California hospital system is being sued by a 33-year laboratory employee who was terminated after missing too many workdays due to “long-haul” COVID-19 illness.
Hamada’s attorney, Amanda Whitten JD of Bryant Whitten LLP in Fresno, told The Fresno Bee that “ [California] state law allows an employee to take up to 12 weeks of leave a year to deal with a serious medical condition,” and that, “It’s also illegal for an employer to retaliate against an employee for requesting and taking that leave.”
Michelle Von Tersch, Senior Vice President of Communications and Legislative Affairs at Community Medical Centers, told the Fresno Bee in a statement that she could not comment on the pending litigation. But she added, “During the COVID-19 pandemic, Community Medical Centers expanded employee assistance programs, including extended time off for employees to care for themselves and their loved ones.”
Community Medical Centers (CMC) is a not-for-profit healthcare system in the greater Fresno area. It operates four hospitals and a cancer institute, and several long-term care, outpatient, and other healthcare facilities. CMC has more than 8,800 employees, according to a hospital fact sheet.
Was Hamada Wrongfully Discharged?
The lawsuit states Hamada worked for Community Hospitals of Central California as a clinical laboratory scientist from July 1, 1987, until Oct. 13, 2020, when she was “wrongfully discharged.” In the filing, Hamada’s attorney noted that Hamada received “good performance reviews and salary increases and was not subject to discipline for her job performance” during her more than 30 years of employment.
After Hamada became sick with COVID-19 in mid-April 2020, she followed her doctor’s recommendation and went on medical leave for roughly six weeks. However, when she returned to work in June 2020, she “still suffered from the effects of the coronavirus” and was considered a “long-haul” COVID-19 patient, the lawsuit states. As a result, her healthcare provider suggested she request “intermittent medical leave” due to her continued illness and underlying medical conditions, including diabetes, cardio-pulmonary disease, and traumatic brain injury.
Plaintiff Alleges Threats and Intimidation
The lawsuit contends Hamada’s request for additional medical leave resulted in her supervisor telling her, “you better not” file the request. In addition to this threat, the plaintiff alleges she was shunned by her supervisor and coworkers and then subjected to discipline based on attendance when she was absent from work due to her medical condition. In October, she was terminated due to violating the “employer’s attendance policy,” the lawsuit states.
The complaint outlines eight causes of action:
Discrimination based on medical condition, disability, or perceived disability.
Failure to accommodate a disability.
Failure to prevent discrimination and discrimination based on medical condition, disability, or perceived disability.
Wrongful termination in violation of public policy.
Defamation.
The California Family Rights Act provides most employees in California with the right to take up to 12 weeks of leave from work to care for themselves or family members with a serious health condition or bond with a new child.
Hamada is requesting a jury award for:
general damages above the jurisdictional minimum of the Court,
special damages,
punitive damages,
interest on lost earnings,
deferred compensation and employee benefits,
reinstatement of her job, and
reimbursement of attorneys’ fees.
Should Long-Haul COVID-19 Be Considered a Disability?
In an article on the Society for Human Resource Management (SHRM) website, David Fram, JD (above), Director of ADA Services with the National Employment Law Institute (NELI), said, “If someone has COVID-19 for two weeks and there are no lingering effects, he or she still could be regarded as having a disability. While an employer doesn’t have to reasonably accommodate someone it merely regards as having a disability, it must refrain from discriminating against that person.” Additionally, he noted, “An employer must not discriminate against and must reasonably accommodate someone who has an impairment that substantially limits a major life activity, which could include a COVID-19 long hauler.” (Photo copyright: National Institutes of Health.)
Are Clinical Laboratories Legally Obligated?
In the same article, S. Leigh Jeter, JD, Senior Counsel with Michael Best and Friedrich in Chicago, said, “Unfortunately, there is no bright-line test for determining whether someone is disabled for purposes of the Act.” She added, “I encourage employers to err on the side of assuming that the employee may be covered under the ADA and then consider those resulting legal obligations.”
Removal of nonessential functions of the position might be a reasonable accommodation, Jeter noted.
Clinical laboratory executives would be wise to follow this COVID-19-related lawsuit closely and review their employment policies to better understand their obligation toward their workers under the Americans with Disabilities Act. This case may open the door to additional lawsuits related to COVID-19 firings.